






最新资讯



-
电话: 400-113-6988 邮箱: dongfangxicao@163.com 地址: 深圳市南山区粤海街道高新中三道9号环球数码大厦19楼
↓ 微信扫码识别关注我们 ↓ 
三氟硒代蛋氨酸:一种新的非天然氨基酸
发表于:2021-07-06 作者:admin 来源:本站 点击量:13246
原文:Block E , Booker S , Flores-Penalba S , et al. Trifluoroselenomethionine: A New Unnatural Amino Acid[J]. Chembiochem, 2016.
翻译:
简介
三氟甲硫氨酸(3,TFM, Scheme 1)和硒甲硫氨酸(5,SeM)是人们感兴趣的前药,分别提供细胞毒性的三氟甲硫氨酸(4)和甲烷硒醇(6)。由蛋氨酸酶(MGL, L-蛋氨酸g-裂解酶,EC 4.4.1.11)裂解的,类似于蛋氨酸(1,Met)的裂解产生甲硫醇(2)。3和5对蛋氨酸酶的Km值明显小于1,这与4和6的酸度高于2相一致。对于4和6的细胞毒性,人们提出了非常不同的机制:4在生理条件下是不稳定的,分解为胺交联剂S=CF2(9,硫碳基氟化),而6产生超氧化物,在氧化谷胱甘肽和其他纤维素硫醇时引发氧化应激。在其他工作中,化合物3被大肠杆菌掺入重组蛋白,提供了19 F NMR光谱探针。它还显示,相对于1,3的硫原子对氧化和与亲电试剂反应的活性降低。
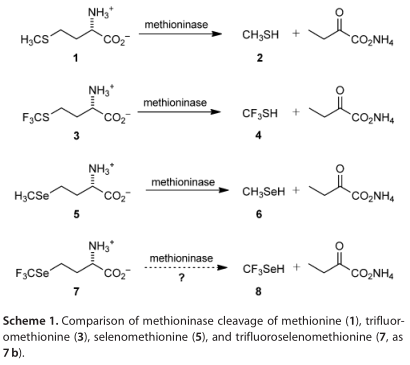
三氟硒代蛋氨酸(7,TFSeM, 硒-三氟-甲基-L-同型半胱氨酸),结合3和5的特征,会表现出相似的生化反应,提供三氟甲烷硒醇(8)。如果是这样,8的细胞毒性与4和6的相比如何?在大肠杆菌中,7是否会和3一样被整合到重组蛋白中,但与5相比,7是否具有更强的抗氧化能力?我们在这里描述的第一个合成7(方案2), 它对甲硫氨酸酶的反应性,氧单电子还原2,6和8的比较计算研究,以及从4和8损失氟离子分别得到硫羰基氟化物(S=CF2,9)和硒羰基氟化物(Se=CF2,10)的比较计算和实验研究。我们还报告了7相对于1、3和5的物理、光谱和计算的结构特性,以及在大肠杆菌中将7并入重组蛋白的努力的结果。
结果与讨论
三氟硒代蛋氨酸的合成
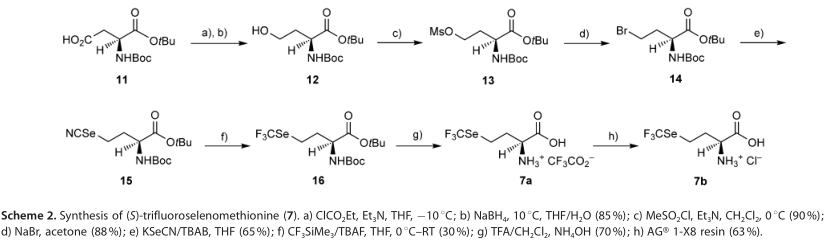
以已知的手性起始物(S)-叔丁基-2-(叔丁氧基羰基氨基)-4-羟基丁酸盐(12,方案2)为原料,合成了一种简便快捷的三氟硒代蛋氨酸(7),化合物12经连续甲磺酸化(化合物13)并经NaBr(化合物14)和KSeCN处理后,得到结晶叔丁基(S)-2-((叔丁氧基羰基)氨基)-4-亚硒酸盐丁酸酯(15,产率约80%),其可由X射线晶体学表征(图1)。高亲脂性的CF3Se组很容易被引入,给出16,以市售三氟甲基三甲基硅烷(CF3SiMe3)与亚硒酸盐15在0~8℃缓慢(注射泵)添加催化(0.2当量)TBAF(四丁基氟化铵)进行处理。亚硒酸盐路线是首选的,因为消耗了所有硒基团。通过用三氟乙酸除去16中的保护基,得到无色固体三氟乙酸盐7a,氟谱的化学位移在-35.12和-76.07,硒谱的化学位移在400ppm。氟谱信号显示烷基取代的CF3Se基团一般出现在-35ppm(相对于CFCl3),与CF3S的-41ppm比较;三氟乙酸出现在-76.55ppm。以AGS 1-X8树脂的氯化物形式处理7a,得到7b,即7的盐酸盐,为无色固体,氟谱的化学位移在-35.35ppm,硒谱的化学位移在400ppm。所有的生物学研究都使用了盐7b。

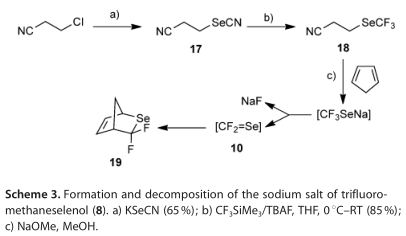
三氟甲烷烯醇(8)的生成与分解
我们试图生成8, 以评估在人结肠癌HCT-116细胞存在下酶促产生的结果。尽管8之前已经被表征过,但据说它在水中不稳定,在加热时分解为HF和F2C=Se。我们试图通过碱催化分解3-(三氟甲基硒基)丙腈(18)来制备8或其钠盐,进而提出用CF3SiMe3处理3-(硒氰酸根)丙腈(17)来制备8或其钠盐(方案3)。在本实验中,17顺利地制备了18,而17又很容易地由3-氯丙腈制备。当用甲醇钠处理18时,用19 F NMR监测反应,仅看到19 F信号对应于氟化物离子,如标准的NMR分析所证实的。当在低温下存在过量的环戊二烯时,用甲醇钠处理18,然后缓慢加热混合物,3,3-二氟-2-硒基环[2.2.1]庚-5-烯(19)-已知的环戊二烯加合物F2C=SE由GC-MS检测,因此,8在溶液中加热后的分解是这样的。
硒代蛋氨酸(5)和三氟硒代蛋氨酸(7)结构的光谱和计算比较
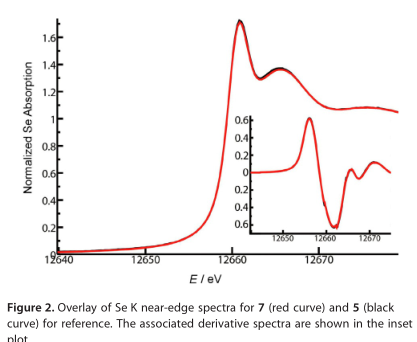
采用光谱和计算相结合的方法,研究了5的取代甲基和7的三氟甲基的物理和电子结构变化。特别是,在斯坦福同步辐射光源(SSRL)的光束线7-3上进行了5和7(as 7b)的扩展X射线吸收精细结构(EXAFS)测量。利用密度泛函理论(DFT)等方法对所得数据进行了分析。Se K近边谱的分析如图2所示,5和7产生了惊人的相似的近边光谱,两者之间的近边没有明显的位移。两个近边谱的一阶导数(图2中的插图)也非常相似,因此不可能通过单独使用Se K近边光谱来区分这两种化学物质。
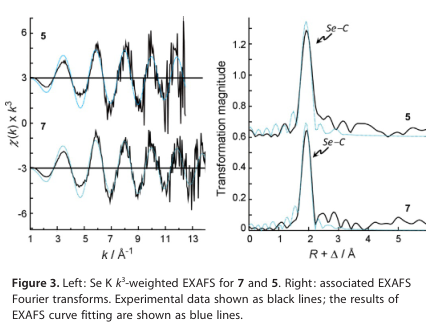
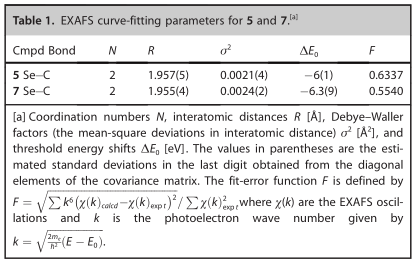
与近边缘谱类似,EXAFS谱和相关的傅里叶变换(图3)对于5和7也非常相似。从EXAFS数据[计算为p/(2Dk),其中Dk是拟合EXAFS数据的范围得到的键长理论分辨率为5 (k范围1-12.5 Å-1)的0.13 Å和7 (k范围1-14 Å-1)的0.12 Å。根据EXAFS数据的键长分辨率,每个类似物中的Se-C键长都非常相似,因此必须将它们分别建模为两个Se-C散射相互作用的单一壳层,从而得到两个相互作用的平均拟合距离(R)。采用EXAFS曲线拟合分析两种Se-C反向散射相互作用的单散射路径,在5和7之间产生了非常相似的键长结果,两个Se-C键分别为1.957(2) Å和两个Se-C键分别为1.955(4) Å (7)(表1)。
在剑桥结构数据库(CSD)中搜索5的Se-C键长,得出Se-CH3的平均原子间距离为(1.944±0.006)Å, Se-CH2的平均原子间距离为(1.954±0.008)Å,略低于从EXAFS数据拟合中获得的平均值(CSD搜索的平均值取自三个小分子晶体结构,每个结构都含有肽链: MAJTAT C-Se-C=97.238°, CH3-Se=1.950, CH2-Se=1.947; DEYFOD C-Se-C=98.688°, CH3-Se =1.939, CH2-Se =1.963; AVIKOG C-Se-C=97.948°, CH3-Se =1.942, CH2-Se =1.952。EXAFS得到的Se-C的平均原子间距仍在CSD搜索Se-CH2键得到的实验误差范围内,仅比CSD搜索Se-CH3的最大值长0.002 Å。
DFT结构计算
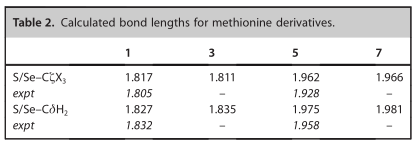
对每个等结构荷电中性氨基酸模型(蛋氨酸(1)和硒代蛋氨酸(5))及其各自含三氟甲基衍生物3和7分别进行了气相几何优化。计算得到的Se-CH3键长为1.96(2) Å,与EXAFS值为1.92(8)a的偏差最大,约为0.03(8) Å。计算出的杂原子-碳键长度(如表2所示)与EXAFS值之间的误差在0.02 Å以内。
DFT计算表明,当甲硫氨酸模型中的末端甲基变成CF3时,硫键长度缩短,同时S-CH2键延伸。类似地,对于含硒类似物,硒-CH2键在含CF3的衍生物中相对于5也被拉长,而硒- CH3和硒- CF3键的长度基本保持不变。基于联合原子拓扑模型,对每一个优化结构进行分子体积计算(图4)。对蛋氨酸侧链的每一个研究修改都会导致侧链体积的增加。
氨基酸5的分子体积比1大4%(如果只考虑杂原子和末端甲基基团,则增加11%)。将1的甲基修饰成CF3,氨基酸体积增加6%(硫和末端基团增加23%),而将5的甲基修饰成CF3则表示分子体积增加了7% (Se-CX3增加了20%)。修改1到7是最重要的增长,相对于蛋氨酸分子体积增加11%,相对于S-CH3增加33%。
相对于1中的硫杂原子,5中的硒原子有较低的d轨道,这比含硫类似物中的硒原子更容易获得能量。结果是,5的LUMO的能量比含硫类似物的能量低,并且更多的轨道密度存在于LUMO中的Se原子上(图4,硫原子处的LUMO密度在使用的轮廓值不明显)。3末端甲基的修饰在杂原子上引入了一个吸电子部分,结果在含硫蛋氨酸类似物中LUMO具有s -c ?*特征。这些趋势在含CF3的硒代蛋氨酸类似物中加倍增强,其中硒原子上电子密度的降低导致LUMO性质的显著改变,使得总密度的显著部分位于杂原子上,尽管在Se K近边谱中,核心1s激发能没有明显的变化(图2),但观察到的C-X-C键角也接近908(图4)。
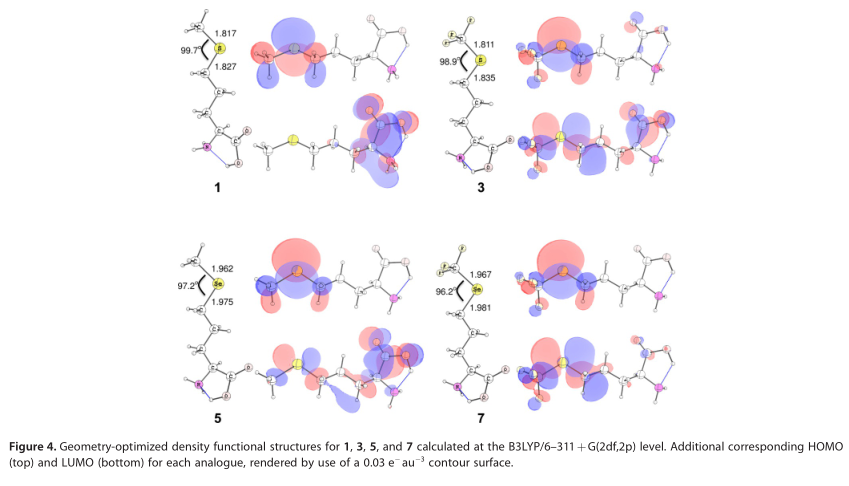
5、3和7的HOMO-LUMO间隙相对于计算出的1的HOMO-LUMO间隙分别为-26.5、+100.9和+32.5 kJ/mol。与含甲基甲硫氨酸残基相比,具有吸电子功能的CF3残基的存在扩大了HOMO-LUMO间隙,而含硫1向含硒5的转变则使HOMO-LUMO间隙变小。7的HOMO-LUMO间隙支持了含硫模型中观察到的CF3修饰的趋势,其中HOMO-LUMO间隙大于含甲基的类似物。
比较7和5的HOMO-LUMO间隙,得出+59.0 kJ/mol的相对变化,再次反映了1和3的趋势。这些数据也汇总在表S2的支持信息中。在3、5和7之间,各种类似物的HOMOs的性质变化相对较小。两种硒代蛋氨酸类似物的荧光性质比较表明,它们在硒原子上都具有一定的p轨道性质。5的LUMO分布在氨基酸中,部分来自Se-C s*和Ca-C(O) p*轨道;然而,在CF3类似物中,Se-C s*轨道对LUMO的性质有重要的贡献,p*对氨基酸主链区域的贡献相对较小。有趣的是,虽然能量不同,3和7的HOMO和LUMO定性相似,尽管3中HOMO-LUMO间隙计算约为70千焦每摩尔(见表S2的支持信息), 与7不同,3的LUMO在主链原子上有更多的p*特征,类似于5(图4)。
Leroux先前断定,过去关于CF3至少和异丙基一样大的断言是不正确的,事实上,CF3的体积比异丙基的体积要小得多。我们还对异丙基和CF3体系进行了模型计算,我们的发现支持了Leroux先前的结论,即CF3取代基确实比CH3大,但体积肯定比异丙基小(数据未显示)。此外,我们还比较了系列S- CH3、S- CF3、Se- CH3和Se- CF3侧链片段的分子空腔。在溶剂化过程中计算出的分子碎片的空腔中包含了溶剂探针分子的额外余量(基于水)。与S- CH3 (47.39Å3)相比,S- CF3的一半体积增大23% (58.43 Å3), Se- CH3的一半体积增大11% (52.60 Å3), Se- CF3的一半体积增大33% (63.22 Å3)。
蛋氨酸酶研究
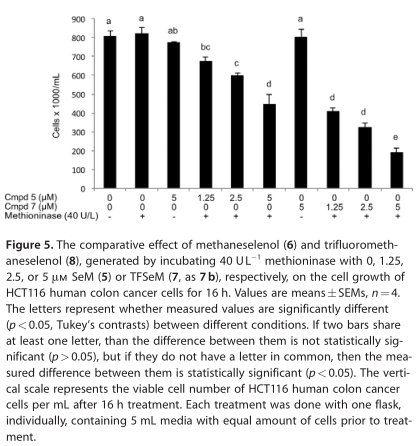
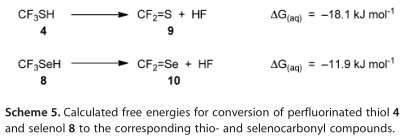
图5显示,经阴道毛滴虫蛋氨酸酶(40 UL -1)、1.25、2.5和处理后,HCT116细胞的生长速度分别受到16.7、26.1和44.6%的抑制,分别用1.25、2.5和5 mmol/L 7b处理的细胞分别为5mmol /L 5、49.4、60.0和76.3%。也就是说,在降低细胞浓度方面,7b比所有测试浓度下的5要有效得多。对于7b相对于5的细胞毒性增强有三种可能的解释,假设蛋氨酸酶从7b释放8,从5释放6:1) CF3SeH(8)比甲烷硒醇更有效地生成活性氧(6,方案4), 2)我们假设CF3SeH(8)在生理条件下会分解为Se=CF2(10),正如我们在模型研究中所做的那样,Se=CF2(10)应该是主要胺基的一个强有力的交联剂,正如所提议的从3到4的细胞毒性,通过形成S=CF2(9,方案5),或者 3)第一和第二假设过程的某些组合发生。
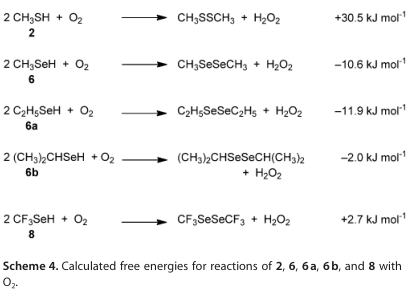
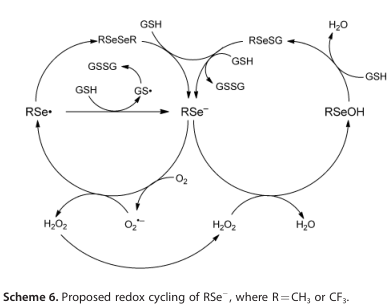
方案6阐述了硒酸盐的氧化还原循环,以及硒酸盐的阴离子形式,如6和8。方案4对比了甲硫醇(2)还原二氧的难易度和硒醇(6,6a, 6b, 8)的DFT计算结果。这些计算表明,硒醇是比硫醇更好的还原剂,6和6a是可比较的(在方法的误差范围内),略优于6b。这些结果与报道的相对细胞毒性6>6a>6b>2的结果一致。方案4的DFT计算并不能正确预测8的相对观察细胞毒性,这一事实表明,需要考虑8的独特性质:例如,氟离子的损失使氟代硒碳酰(方案5中计算的10,之前报道的4 [1a]),正如我们经实验证实为8。第三种可能性是,与8有关的细胞毒性是由于它的酸性增强(如方案5所计算)或其相应阴离子的氧化还原特性。报告中的的2、4、6和8的pKa值分别为10.09±0.10、2.77±0.10、6.50±0.70和-1.29±0.70。
TFSeM与大肠杆菌蛋白的结合
将含氟氨基酸结合到蛋白质中,提供了一种结构探针,并有可能引入有用的功能变化。甲硫氨酸tRNA合成酶(MetRS)是一种比较混杂的氨酰tRNA合成酶,但它仍然根据侧链的长度和几何结构行使选择性。鉴于二氟甲硫氨酸和三氟甲硫氨酸(3)已成功地结合到蛋白质中,因此确定TFSeM(7)是否可以类似于合并。用含氟类似物替代特定氨基酸的标准程序包括在无法合成传统氨基酸的大肠杆菌辅助营养体中表达目标重组蛋白。将氟化氨基酸添加到限定的培养基中,由细胞吸收,然后并入蛋白质中。成功的取代既依赖于所讨论的氨基酰核糖核酸合成酶利用氟化类似物的能力,也依赖于当插入类似物时新生链折叠成稳定三维结构的能力。通过这一程序实现的合并比率往往很低,由于在将氟化氨基酸装入特定的氨基酰核糖核酸合成酶方面存在挑战,以前的文献报道了10-15%的成功将3并入绿色荧光蛋白,31-70%的成功将3并入噬菌体l溶菌酶的不同位置。这些研究表明,氟化氨基酸的掺入对于暴露在溶剂中的位置是最有效的,在这些位置,更大的体积和疏水性的变化可以更好地耐受。
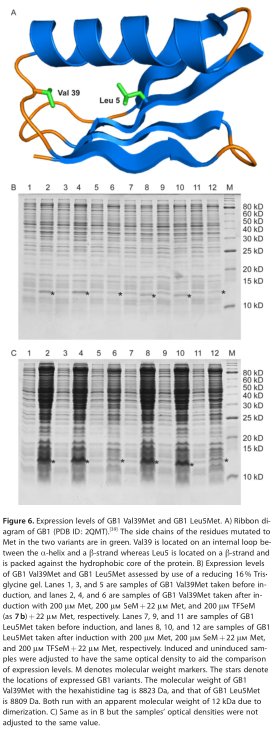
为了评估7的掺入,我们选择链球菌蛋白G(G B1)的56个氨基酸蛋白免疫球蛋白结合B1结构域作为靶点。GB1具有非常稳定的折叠,经常被用于蛋白质折叠和热稳定性的研究。我们构建了一个在纯化后,每个蛋白中都有一个单独的Met残基存在于蛋白质中的变体:一个Val39Met变体,其中Met位于环中;一个Leu5Met变体,其中Met位于环中位于蛋白质的疏水核心(图6A)。相似的替换被证明是稳定的和折叠良好的。比较两个不同变体的表达水平可以检验7的结合是否是位置依赖的。
我们遵循报告的表达程序,加入3和其他蛋氨酸类似物,并进行以下修改。首先,我们利用C43(DE3)为基础的大肠杆菌氨基酸营养缺陷型宿主菌株RF11,其中Meta被删除,其次,MET被包含在生长培养基中,因为先前的研究表明,在生长培养基中包含10%的Met可以降低SEM的毒性,同时允许高的SEM掺入比。为了减少7的毒性,以其盐酸盐7B的形式,Met营养缺陷型RF11在溶生性培养液(LB)中生长,并在生长的中对数阶段切换到定义的培养基中。以9:1摩尔比添加Met、SeM和Met,或以9:1摩尔比添加TFSeM(as 7b)和Met。通过SDS-PAGE分析比较表达水平(图6B和C)。在TFSeM(AS 7b)存在下生长的GB1的表达水平低于在单独存在或SEM和MET存在下检测到的,从而表明TFSeM比SEM更具细胞毒性,与先前关于氟化氨基酸毒性的报告一致。
为测定TFSeM (as 7b)的掺入率,用IMAC层析法对两种蛋白进行纯化,实验部分详细介绍。纯化后的GB1 Leu5Met和GB1 Val39Met的质谱中,以9:1摩尔比的TFSeM (as 7b)/Met生长,没有明显的TFSeM插入的证据(图7b和D),相反,蛋白质的分子质量表明加入了SeM。当细胞在9:1摩尔比的SeM/Met条件下生长时,GB1 Leu5Met和GB1 Val39Met主要含有SeM,这反映了其相对于所定义的培养基中Met的丰度(图7A和C)。
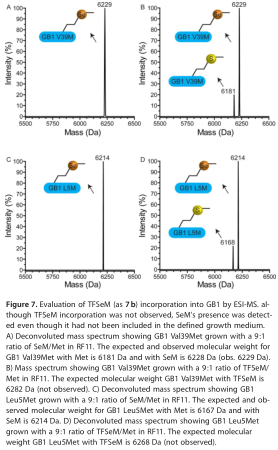
由于在Met auxotroph的生长培养基中既没有SeM也没有其他硒源,如果SeM确实与GB1结合,那么插入GB1的观察到的SeM一定来源于TFSeM。为了确认ESI-MS检测到的蛋白种类,我们对RF11中制备的GB1 Val39Met进行了放大。通过完整蛋白质ESI-MS和串联质谱对所得样品进行分析。在生长培养基中,分别用22 mm Met(图8A)、22 mm Met和200 mm TFSeM(图7b、图8B)和200 mm TFSeM(图7b、图8C)过度生产了GB1 Val39Met。本实验观察到的主要蛋白形态为GB1 al39Met(计算分子量为6179 Da,观察分子量为6180 Da)。也存在低分子量的形式,可能来自于蛋白质中一个甲基(6166 Da)或水(6166 Da)的损失。这些变化可能是由于在低Met浓度下生长的Met生长抑菌所产生的应力所致。在6220/1 Da处形成的蛋白很可能是体内乙酰化或与LC中使用的乙腈相互作用的结果。只有当生长培养基中使用22mm Met和200mm TFSeM过量产生GB1 Val39Met时,才有可能检测到怀疑含有SeM的6227 Da峰。图8B为经胰酶消化后串联ms测序的样品。图8D为肽QYANDNG(SeM 39)GDEWTYDDATK,根据其分子量和硒的不同同位素模式进行鉴定。由于该肽在样品中的丰度较低,因此无法对其进行完全测序。有趣的是,尽管该样品还含有一种6280 Da的物质,其分子量与计算出的GB1 Val39TFSeM相匹配,未检测到相应的肽QYANDNG(TFSeM 39)GDEWTYDDATK。因此,我们无法从实验上证实TFSeM在这些特定的生长条件下被纳入GB1。
如前所述,由于生长培养基中既没有SeM也没有其他硒来源,且在生长培养基中没有TFSeM而过量产生GB1 Val39Met时没有观察到SeM蛋白,因此我们推断TFSeM片段在体内还原为SeM。在TFSeM存在下培养的大肠杆菌中,SeM在GB1 Val39Met和Leu5Met中表达,可以设想通过中间形成TFSeSAM来实现。在这种途径中,一个或多个依赖于s -腺苷蛋氨酸(SAM)的甲基转移酶(MTases)可将三氟甲基基移除,这些甲基转移酶通常将SAM的甲基部分转移到目标受体(方案7)。由此产生的化合物,se -腺苷基同型半胱氨酸(SeAH),将会被s -腺苷同型半胱氨酸/甲基硫代腺苷核苷酶生成腺嘌呤和se -核糖基-同型半胱氨酸,后者通过核糖体同型半胱氨酸裂解酶转化为同型半胱氨酸和(4S)-4,5-二羟基戊烷-2,3-二酮。然后,高半胱氨酸被依赖于钴胺或不依赖于钴胺的蛋氨酸合酶甲基化,生成硒代蛋氨酸。同型半胱氨酸的甲基化也可以通过不太为人所知的机制发生,如mmuM基因在大肠杆菌中产生的同型半胱氨酸S-甲基转移酶的作用,它使用l-S-甲基蛋氨酸或SAM的S(+ +)异构体作为甲基供体。
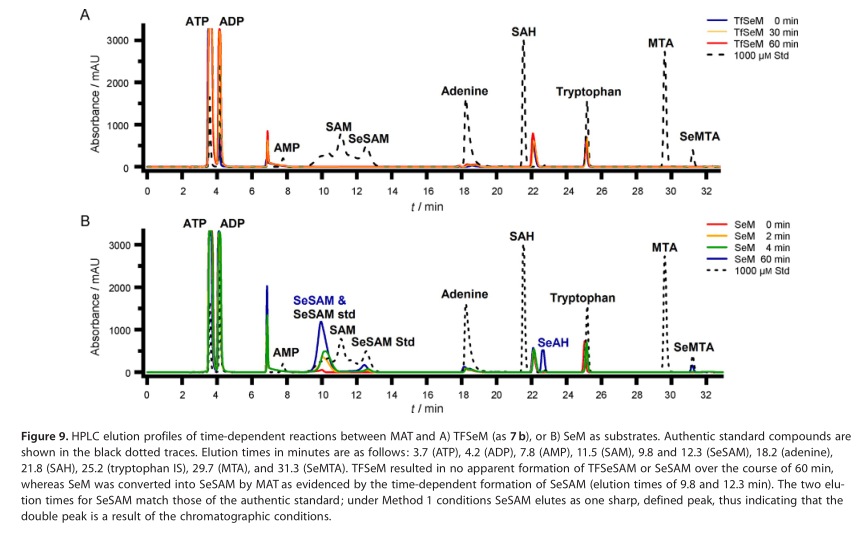
为了确定TFSeSAM是否是由TFSeM形成SeM的关键中间体,对大肠杆菌甲硫氨酸腺苷转移酶(MAT)将TFSeM转化为TFSeSAM的能力进行了评估。MAT催化L-蛋氨酸和ATP缩合形成SAM,并作为副产物生成焦磷酸盐和无机磷酸盐。图9A显示了在有TFSeM(as 7b)的情况下,在25°C的转换条件下培养30或60分钟的MAT反应产物的HPLC洗脱曲线。含有相关腺嘌呤化合物(虚线)和色氨酸(用作内标)的真实标准的痕量表明,在11到13分钟之间洗脱SAM和SeSAM。在30和60分钟时间点,在含有TFSeM的反应中,SAM或SeSAM的对应区域均不存在峰,整个色谱图中的可见峰在反应开始前(0分钟)出现,且不随时间增加(蓝、黄和红线)。在相同条件下进行的对照反应的高效液相色谱洗脱谱(但包含SeM而非TFSeM)显示,SeSAM的形成稳健且随时间变化(图9B,红色、黄色、绿色和蓝色的痕迹),表明MAT制剂是活性的。SAH之后未知的洗脱峰也出现在TFSeM反应中,很可能是MAT制备过程中的污染物。SeAH的观察表明,MAT制剂也被一个或多个甲基转移酶污染。

用LC-MS分析类似的反应,以评估TFSeM (as 7b)的浓度是否随时间变化。图S1A显示了在SeM存在下不同孵育程度下MAT反应的总离子色谱图。峰值在1.14、1.39、1.54和4.46分钟出现随时间变化。在1.39 min (m/z 198.1)时,其峰值与SeM相对应,并随时间变化而降低,而在1.14 min (m/z 447.1)、1.54 min (m/z 433.1)和4.46 min (m/z 346.1)时,其峰值分别与SeSAM、SeAH和硒腺苷(MSeA)相对应,并随时间变化而升高。SeAH和MSeA的形成是意料之中的,考虑到MAT制备过程中的甲基转移酶污染以及SeSAM非酶催化形成MSeA和同丝氨酸内酯的倾向。相比之下,在不同程度的TFSeM孵育下,MAT反应的总离子色谱图显示,在孵育18小时后,TFSeM的浓度变化不明显(图S1B)。
鉴于大肠杆菌MAT似乎不使用TFSeM作为底物,我们进行了实验,以评估大肠杆菌中的其他蛋白质是否可以作用于它。在这些实验中,用大肠杆菌蛋氨酸营养不良菌株B834(DE3)pLysS的粗裂解液对1 mm的TFSeM(as 7b)进行不同时间的孵育,并用LC-MS分析反应。如图10A和B所示,TFSeM和SeM均以时间依赖的方式消耗。当用TFSeM和粗裂解液进行类似反应时煮沸10分钟或在含有分子量为3 kDa的膜的旋转浓缩器中离心的粗裂解液滤液,在整个18小时的培养期内,SeM和TFSeM的浓度保持恒定(图S2A-D)。这种行为强烈地表明,TFSeM在大肠杆菌中是由一种未经鉴定的蛋白质作用的。总之,这些结果表明,尽管TFSeM未转化为TFSeSAM,但它是大肠杆菌中另一种蛋白质的靶点,最有可能将其转化为高硒半胱氨酸或该代谢物的前体。
结论
以N-(叔丁基羰基)-L-天冬氨酸叔丁基酯为原料,经七步反应合成了一种新的非天然氨基酸三氟硒代蛋氨酸(7)。与硒代蛋氨酸(5)相比,三氟硒代蛋氨酸对人结肠癌HCT-116细胞的细胞毒性增强。通过计算和实验验证了这种增强活性的机制解释。用EXAFS和DFT计算5和7的结果表明,它们在光谱和结构上非常相似。然而,当用7(as 7b)和蛋氨酸以9:1的比例在大肠杆菌蛋氨酸营养不良细胞系RF11中表达两个不同的蛋白GB1突变体时,发现令人惊讶的是,超过80%的蛋白含有5,即使没有添加5,这意味着三氟甲基从7失去。一种未知的酶催化这个过程。确定是哪一种酶导致了7的三氟甲基的缺失是很有意义的,因为这种化学基团存在于许多药物中,因此具有内在的药用价值。
翻译:
三氟硒代蛋氨酸:一种新的非天然氨基酸
以N-(叔丁基丁基苯基)-L-天冬氨酸叔丁基酯为原料,经七步反应合成了一种新的非天然氨基酸三氟硒代蛋氨酸(TFSeM)。TFSeM显示,与硒代蛋氨酸(SeM)相比,蛋氨酸酶对人结肠癌HCT-116细胞的细胞毒性增强。这种增强活性的机制解释是通过计算和实验验证的。用EXAFS和DFT对TFSeM和SeM进行了比较,结果表明两者在光谱和结构上非常相似。尽管如此,当在大肠杆菌蛋氨酸营养不良细胞系中以9:1的摩尔比在TFSeM和蛋氨酸(Met)存在下表达两种不同的蛋白GB1时,令人惊讶的是,85%的蛋白质含有SeM残基,尽管没有添加SeM,这意味着三氟甲基在TFSeM中的损失。大肠杆菌提取物酶促TFSeM向SeM转化,但TFSeM不是大肠杆菌蛋氨酸腺苷转移酶的底物。简介
三氟甲硫氨酸(3,TFM, Scheme 1)和硒甲硫氨酸(5,SeM)是人们感兴趣的前药,分别提供细胞毒性的三氟甲硫氨酸(4)和甲烷硒醇(6)。由蛋氨酸酶(MGL, L-蛋氨酸g-裂解酶,EC 4.4.1.11)裂解的,类似于蛋氨酸(1,Met)的裂解产生甲硫醇(2)。3和5对蛋氨酸酶的Km值明显小于1,这与4和6的酸度高于2相一致。对于4和6的细胞毒性,人们提出了非常不同的机制:4在生理条件下是不稳定的,分解为胺交联剂S=CF2(9,硫碳基氟化),而6产生超氧化物,在氧化谷胱甘肽和其他纤维素硫醇时引发氧化应激。在其他工作中,化合物3被大肠杆菌掺入重组蛋白,提供了19 F NMR光谱探针。它还显示,相对于1,3的硫原子对氧化和与亲电试剂反应的活性降低。
三氟硒代蛋氨酸(7,TFSeM, 硒-三氟-甲基-L-同型半胱氨酸),结合3和5的特征,会表现出相似的生化反应,提供三氟甲烷硒醇(8)。如果是这样,8的细胞毒性与4和6的相比如何?在大肠杆菌中,7是否会和3一样被整合到重组蛋白中,但与5相比,7是否具有更强的抗氧化能力?我们在这里描述的第一个合成7(方案2), 它对甲硫氨酸酶的反应性,氧单电子还原2,6和8的比较计算研究,以及从4和8损失氟离子分别得到硫羰基氟化物(S=CF2,9)和硒羰基氟化物(Se=CF2,10)的比较计算和实验研究。我们还报告了7相对于1、3和5的物理、光谱和计算的结构特性,以及在大肠杆菌中将7并入重组蛋白的努力的结果。
结果与讨论
三氟硒代蛋氨酸的合成
以已知的手性起始物(S)-叔丁基-2-(叔丁氧基羰基氨基)-4-羟基丁酸盐(12,方案2)为原料,合成了一种简便快捷的三氟硒代蛋氨酸(7),化合物12经连续甲磺酸化(化合物13)并经NaBr(化合物14)和KSeCN处理后,得到结晶叔丁基(S)-2-((叔丁氧基羰基)氨基)-4-亚硒酸盐丁酸酯(15,产率约80%),其可由X射线晶体学表征(图1)。高亲脂性的CF3Se组很容易被引入,给出16,以市售三氟甲基三甲基硅烷(CF3SiMe3)与亚硒酸盐15在0~8℃缓慢(注射泵)添加催化(0.2当量)TBAF(四丁基氟化铵)进行处理。亚硒酸盐路线是首选的,因为消耗了所有硒基团。通过用三氟乙酸除去16中的保护基,得到无色固体三氟乙酸盐7a,氟谱的化学位移在-35.12和-76.07,硒谱的化学位移在400ppm。氟谱信号显示烷基取代的CF3Se基团一般出现在-35ppm(相对于CFCl3),与CF3S的-41ppm比较;三氟乙酸出现在-76.55ppm。以AGS 1-X8树脂的氯化物形式处理7a,得到7b,即7的盐酸盐,为无色固体,氟谱的化学位移在-35.35ppm,硒谱的化学位移在400ppm。所有的生物学研究都使用了盐7b。
三氟甲烷烯醇(8)的生成与分解
我们试图生成8, 以评估在人结肠癌HCT-116细胞存在下酶促产生的结果。尽管8之前已经被表征过,但据说它在水中不稳定,在加热时分解为HF和F2C=Se。我们试图通过碱催化分解3-(三氟甲基硒基)丙腈(18)来制备8或其钠盐,进而提出用CF3SiMe3处理3-(硒氰酸根)丙腈(17)来制备8或其钠盐(方案3)。在本实验中,17顺利地制备了18,而17又很容易地由3-氯丙腈制备。当用甲醇钠处理18时,用19 F NMR监测反应,仅看到19 F信号对应于氟化物离子,如标准的NMR分析所证实的。当在低温下存在过量的环戊二烯时,用甲醇钠处理18,然后缓慢加热混合物,3,3-二氟-2-硒基环[2.2.1]庚-5-烯(19)-已知的环戊二烯加合物F2C=SE由GC-MS检测,因此,8在溶液中加热后的分解是这样的。
硒代蛋氨酸(5)和三氟硒代蛋氨酸(7)结构的光谱和计算比较
采用光谱和计算相结合的方法,研究了5的取代甲基和7的三氟甲基的物理和电子结构变化。特别是,在斯坦福同步辐射光源(SSRL)的光束线7-3上进行了5和7(as 7b)的扩展X射线吸收精细结构(EXAFS)测量。利用密度泛函理论(DFT)等方法对所得数据进行了分析。Se K近边谱的分析如图2所示,5和7产生了惊人的相似的近边光谱,两者之间的近边没有明显的位移。两个近边谱的一阶导数(图2中的插图)也非常相似,因此不可能通过单独使用Se K近边光谱来区分这两种化学物质。
与近边缘谱类似,EXAFS谱和相关的傅里叶变换(图3)对于5和7也非常相似。从EXAFS数据[计算为p/(2Dk),其中Dk是拟合EXAFS数据的范围得到的键长理论分辨率为5 (k范围1-12.5 Å-1)的0.13 Å和7 (k范围1-14 Å-1)的0.12 Å。根据EXAFS数据的键长分辨率,每个类似物中的Se-C键长都非常相似,因此必须将它们分别建模为两个Se-C散射相互作用的单一壳层,从而得到两个相互作用的平均拟合距离(R)。采用EXAFS曲线拟合分析两种Se-C反向散射相互作用的单散射路径,在5和7之间产生了非常相似的键长结果,两个Se-C键分别为1.957(2) Å和两个Se-C键分别为1.955(4) Å (7)(表1)。
在剑桥结构数据库(CSD)中搜索5的Se-C键长,得出Se-CH3的平均原子间距离为(1.944±0.006)Å, Se-CH2的平均原子间距离为(1.954±0.008)Å,略低于从EXAFS数据拟合中获得的平均值(CSD搜索的平均值取自三个小分子晶体结构,每个结构都含有肽链: MAJTAT C-Se-C=97.238°, CH3-Se=1.950, CH2-Se=1.947; DEYFOD C-Se-C=98.688°, CH3-Se =1.939, CH2-Se =1.963; AVIKOG C-Se-C=97.948°, CH3-Se =1.942, CH2-Se =1.952。EXAFS得到的Se-C的平均原子间距仍在CSD搜索Se-CH2键得到的实验误差范围内,仅比CSD搜索Se-CH3的最大值长0.002 Å。
DFT结构计算
对每个等结构荷电中性氨基酸模型(蛋氨酸(1)和硒代蛋氨酸(5))及其各自含三氟甲基衍生物3和7分别进行了气相几何优化。计算得到的Se-CH3键长为1.96(2) Å,与EXAFS值为1.92(8)a的偏差最大,约为0.03(8) Å。计算出的杂原子-碳键长度(如表2所示)与EXAFS值之间的误差在0.02 Å以内。
DFT计算表明,当甲硫氨酸模型中的末端甲基变成CF3时,硫键长度缩短,同时S-CH2键延伸。类似地,对于含硒类似物,硒-CH2键在含CF3的衍生物中相对于5也被拉长,而硒- CH3和硒- CF3键的长度基本保持不变。基于联合原子拓扑模型,对每一个优化结构进行分子体积计算(图4)。对蛋氨酸侧链的每一个研究修改都会导致侧链体积的增加。
氨基酸5的分子体积比1大4%(如果只考虑杂原子和末端甲基基团,则增加11%)。将1的甲基修饰成CF3,氨基酸体积增加6%(硫和末端基团增加23%),而将5的甲基修饰成CF3则表示分子体积增加了7% (Se-CX3增加了20%)。修改1到7是最重要的增长,相对于蛋氨酸分子体积增加11%,相对于S-CH3增加33%。
相对于1中的硫杂原子,5中的硒原子有较低的d轨道,这比含硫类似物中的硒原子更容易获得能量。结果是,5的LUMO的能量比含硫类似物的能量低,并且更多的轨道密度存在于LUMO中的Se原子上(图4,硫原子处的LUMO密度在使用的轮廓值不明显)。3末端甲基的修饰在杂原子上引入了一个吸电子部分,结果在含硫蛋氨酸类似物中LUMO具有s -c ?*特征。这些趋势在含CF3的硒代蛋氨酸类似物中加倍增强,其中硒原子上电子密度的降低导致LUMO性质的显著改变,使得总密度的显著部分位于杂原子上,尽管在Se K近边谱中,核心1s激发能没有明显的变化(图2),但观察到的C-X-C键角也接近908(图4)。
5、3和7的HOMO-LUMO间隙相对于计算出的1的HOMO-LUMO间隙分别为-26.5、+100.9和+32.5 kJ/mol。与含甲基甲硫氨酸残基相比,具有吸电子功能的CF3残基的存在扩大了HOMO-LUMO间隙,而含硫1向含硒5的转变则使HOMO-LUMO间隙变小。7的HOMO-LUMO间隙支持了含硫模型中观察到的CF3修饰的趋势,其中HOMO-LUMO间隙大于含甲基的类似物。
比较7和5的HOMO-LUMO间隙,得出+59.0 kJ/mol的相对变化,再次反映了1和3的趋势。这些数据也汇总在表S2的支持信息中。在3、5和7之间,各种类似物的HOMOs的性质变化相对较小。两种硒代蛋氨酸类似物的荧光性质比较表明,它们在硒原子上都具有一定的p轨道性质。5的LUMO分布在氨基酸中,部分来自Se-C s*和Ca-C(O) p*轨道;然而,在CF3类似物中,Se-C s*轨道对LUMO的性质有重要的贡献,p*对氨基酸主链区域的贡献相对较小。有趣的是,虽然能量不同,3和7的HOMO和LUMO定性相似,尽管3中HOMO-LUMO间隙计算约为70千焦每摩尔(见表S2的支持信息), 与7不同,3的LUMO在主链原子上有更多的p*特征,类似于5(图4)。
Leroux先前断定,过去关于CF3至少和异丙基一样大的断言是不正确的,事实上,CF3的体积比异丙基的体积要小得多。我们还对异丙基和CF3体系进行了模型计算,我们的发现支持了Leroux先前的结论,即CF3取代基确实比CH3大,但体积肯定比异丙基小(数据未显示)。此外,我们还比较了系列S- CH3、S- CF3、Se- CH3和Se- CF3侧链片段的分子空腔。在溶剂化过程中计算出的分子碎片的空腔中包含了溶剂探针分子的额外余量(基于水)。与S- CH3 (47.39Å3)相比,S- CF3的一半体积增大23% (58.43 Å3), Se- CH3的一半体积增大11% (52.60 Å3), Se- CF3的一半体积增大33% (63.22 Å3)。
蛋氨酸酶研究
图5显示,经阴道毛滴虫蛋氨酸酶(40 UL -1)、1.25、2.5和处理后,HCT116细胞的生长速度分别受到16.7、26.1和44.6%的抑制,分别用1.25、2.5和5 mmol/L 7b处理的细胞分别为5mmol /L 5、49.4、60.0和76.3%。也就是说,在降低细胞浓度方面,7b比所有测试浓度下的5要有效得多。对于7b相对于5的细胞毒性增强有三种可能的解释,假设蛋氨酸酶从7b释放8,从5释放6:1) CF3SeH(8)比甲烷硒醇更有效地生成活性氧(6,方案4), 2)我们假设CF3SeH(8)在生理条件下会分解为Se=CF2(10),正如我们在模型研究中所做的那样,Se=CF2(10)应该是主要胺基的一个强有力的交联剂,正如所提议的从3到4的细胞毒性,通过形成S=CF2(9,方案5),或者 3)第一和第二假设过程的某些组合发生。
方案6阐述了硒酸盐的氧化还原循环,以及硒酸盐的阴离子形式,如6和8。方案4对比了甲硫醇(2)还原二氧的难易度和硒醇(6,6a, 6b, 8)的DFT计算结果。这些计算表明,硒醇是比硫醇更好的还原剂,6和6a是可比较的(在方法的误差范围内),略优于6b。这些结果与报道的相对细胞毒性6>6a>6b>2的结果一致。方案4的DFT计算并不能正确预测8的相对观察细胞毒性,这一事实表明,需要考虑8的独特性质:例如,氟离子的损失使氟代硒碳酰(方案5中计算的10,之前报道的4 [1a]),正如我们经实验证实为8。第三种可能性是,与8有关的细胞毒性是由于它的酸性增强(如方案5所计算)或其相应阴离子的氧化还原特性。报告中的的2、4、6和8的pKa值分别为10.09±0.10、2.77±0.10、6.50±0.70和-1.29±0.70。
TFSeM与大肠杆菌蛋白的结合
将含氟氨基酸结合到蛋白质中,提供了一种结构探针,并有可能引入有用的功能变化。甲硫氨酸tRNA合成酶(MetRS)是一种比较混杂的氨酰tRNA合成酶,但它仍然根据侧链的长度和几何结构行使选择性。鉴于二氟甲硫氨酸和三氟甲硫氨酸(3)已成功地结合到蛋白质中,因此确定TFSeM(7)是否可以类似于合并。用含氟类似物替代特定氨基酸的标准程序包括在无法合成传统氨基酸的大肠杆菌辅助营养体中表达目标重组蛋白。将氟化氨基酸添加到限定的培养基中,由细胞吸收,然后并入蛋白质中。成功的取代既依赖于所讨论的氨基酰核糖核酸合成酶利用氟化类似物的能力,也依赖于当插入类似物时新生链折叠成稳定三维结构的能力。通过这一程序实现的合并比率往往很低,由于在将氟化氨基酸装入特定的氨基酰核糖核酸合成酶方面存在挑战,以前的文献报道了10-15%的成功将3并入绿色荧光蛋白,31-70%的成功将3并入噬菌体l溶菌酶的不同位置。这些研究表明,氟化氨基酸的掺入对于暴露在溶剂中的位置是最有效的,在这些位置,更大的体积和疏水性的变化可以更好地耐受。
为了评估7的掺入,我们选择链球菌蛋白G(G B1)的56个氨基酸蛋白免疫球蛋白结合B1结构域作为靶点。GB1具有非常稳定的折叠,经常被用于蛋白质折叠和热稳定性的研究。我们构建了一个在纯化后,每个蛋白中都有一个单独的Met残基存在于蛋白质中的变体:一个Val39Met变体,其中Met位于环中;一个Leu5Met变体,其中Met位于环中位于蛋白质的疏水核心(图6A)。相似的替换被证明是稳定的和折叠良好的。比较两个不同变体的表达水平可以检验7的结合是否是位置依赖的。
我们遵循报告的表达程序,加入3和其他蛋氨酸类似物,并进行以下修改。首先,我们利用C43(DE3)为基础的大肠杆菌氨基酸营养缺陷型宿主菌株RF11,其中Meta被删除,其次,MET被包含在生长培养基中,因为先前的研究表明,在生长培养基中包含10%的Met可以降低SEM的毒性,同时允许高的SEM掺入比。为了减少7的毒性,以其盐酸盐7B的形式,Met营养缺陷型RF11在溶生性培养液(LB)中生长,并在生长的中对数阶段切换到定义的培养基中。以9:1摩尔比添加Met、SeM和Met,或以9:1摩尔比添加TFSeM(as 7b)和Met。通过SDS-PAGE分析比较表达水平(图6B和C)。在TFSeM(AS 7b)存在下生长的GB1的表达水平低于在单独存在或SEM和MET存在下检测到的,从而表明TFSeM比SEM更具细胞毒性,与先前关于氟化氨基酸毒性的报告一致。
为测定TFSeM (as 7b)的掺入率,用IMAC层析法对两种蛋白进行纯化,实验部分详细介绍。纯化后的GB1 Leu5Met和GB1 Val39Met的质谱中,以9:1摩尔比的TFSeM (as 7b)/Met生长,没有明显的TFSeM插入的证据(图7b和D),相反,蛋白质的分子质量表明加入了SeM。当细胞在9:1摩尔比的SeM/Met条件下生长时,GB1 Leu5Met和GB1 Val39Met主要含有SeM,这反映了其相对于所定义的培养基中Met的丰度(图7A和C)。
由于在Met auxotroph的生长培养基中既没有SeM也没有其他硒源,如果SeM确实与GB1结合,那么插入GB1的观察到的SeM一定来源于TFSeM。为了确认ESI-MS检测到的蛋白种类,我们对RF11中制备的GB1 Val39Met进行了放大。通过完整蛋白质ESI-MS和串联质谱对所得样品进行分析。在生长培养基中,分别用22 mm Met(图8A)、22 mm Met和200 mm TFSeM(图7b、图8B)和200 mm TFSeM(图7b、图8C)过度生产了GB1 Val39Met。本实验观察到的主要蛋白形态为GB1 al39Met(计算分子量为6179 Da,观察分子量为6180 Da)。也存在低分子量的形式,可能来自于蛋白质中一个甲基(6166 Da)或水(6166 Da)的损失。这些变化可能是由于在低Met浓度下生长的Met生长抑菌所产生的应力所致。在6220/1 Da处形成的蛋白很可能是体内乙酰化或与LC中使用的乙腈相互作用的结果。只有当生长培养基中使用22mm Met和200mm TFSeM过量产生GB1 Val39Met时,才有可能检测到怀疑含有SeM的6227 Da峰。图8B为经胰酶消化后串联ms测序的样品。图8D为肽QYANDNG(SeM 39)GDEWTYDDATK,根据其分子量和硒的不同同位素模式进行鉴定。由于该肽在样品中的丰度较低,因此无法对其进行完全测序。有趣的是,尽管该样品还含有一种6280 Da的物质,其分子量与计算出的GB1 Val39TFSeM相匹配,未检测到相应的肽QYANDNG(TFSeM 39)GDEWTYDDATK。因此,我们无法从实验上证实TFSeM在这些特定的生长条件下被纳入GB1。
如前所述,由于生长培养基中既没有SeM也没有其他硒来源,且在生长培养基中没有TFSeM而过量产生GB1 Val39Met时没有观察到SeM蛋白,因此我们推断TFSeM片段在体内还原为SeM。在TFSeM存在下培养的大肠杆菌中,SeM在GB1 Val39Met和Leu5Met中表达,可以设想通过中间形成TFSeSAM来实现。在这种途径中,一个或多个依赖于s -腺苷蛋氨酸(SAM)的甲基转移酶(MTases)可将三氟甲基基移除,这些甲基转移酶通常将SAM的甲基部分转移到目标受体(方案7)。由此产生的化合物,se -腺苷基同型半胱氨酸(SeAH),将会被s -腺苷同型半胱氨酸/甲基硫代腺苷核苷酶生成腺嘌呤和se -核糖基-同型半胱氨酸,后者通过核糖体同型半胱氨酸裂解酶转化为同型半胱氨酸和(4S)-4,5-二羟基戊烷-2,3-二酮。然后,高半胱氨酸被依赖于钴胺或不依赖于钴胺的蛋氨酸合酶甲基化,生成硒代蛋氨酸。同型半胱氨酸的甲基化也可以通过不太为人所知的机制发生,如mmuM基因在大肠杆菌中产生的同型半胱氨酸S-甲基转移酶的作用,它使用l-S-甲基蛋氨酸或SAM的S(+ +)异构体作为甲基供体。
为了确定TFSeSAM是否是由TFSeM形成SeM的关键中间体,对大肠杆菌甲硫氨酸腺苷转移酶(MAT)将TFSeM转化为TFSeSAM的能力进行了评估。MAT催化L-蛋氨酸和ATP缩合形成SAM,并作为副产物生成焦磷酸盐和无机磷酸盐。图9A显示了在有TFSeM(as 7b)的情况下,在25°C的转换条件下培养30或60分钟的MAT反应产物的HPLC洗脱曲线。含有相关腺嘌呤化合物(虚线)和色氨酸(用作内标)的真实标准的痕量表明,在11到13分钟之间洗脱SAM和SeSAM。在30和60分钟时间点,在含有TFSeM的反应中,SAM或SeSAM的对应区域均不存在峰,整个色谱图中的可见峰在反应开始前(0分钟)出现,且不随时间增加(蓝、黄和红线)。在相同条件下进行的对照反应的高效液相色谱洗脱谱(但包含SeM而非TFSeM)显示,SeSAM的形成稳健且随时间变化(图9B,红色、黄色、绿色和蓝色的痕迹),表明MAT制剂是活性的。SAH之后未知的洗脱峰也出现在TFSeM反应中,很可能是MAT制备过程中的污染物。SeAH的观察表明,MAT制剂也被一个或多个甲基转移酶污染。
用LC-MS分析类似的反应,以评估TFSeM (as 7b)的浓度是否随时间变化。图S1A显示了在SeM存在下不同孵育程度下MAT反应的总离子色谱图。峰值在1.14、1.39、1.54和4.46分钟出现随时间变化。在1.39 min (m/z 198.1)时,其峰值与SeM相对应,并随时间变化而降低,而在1.14 min (m/z 447.1)、1.54 min (m/z 433.1)和4.46 min (m/z 346.1)时,其峰值分别与SeSAM、SeAH和硒腺苷(MSeA)相对应,并随时间变化而升高。SeAH和MSeA的形成是意料之中的,考虑到MAT制备过程中的甲基转移酶污染以及SeSAM非酶催化形成MSeA和同丝氨酸内酯的倾向。相比之下,在不同程度的TFSeM孵育下,MAT反应的总离子色谱图显示,在孵育18小时后,TFSeM的浓度变化不明显(图S1B)。
鉴于大肠杆菌MAT似乎不使用TFSeM作为底物,我们进行了实验,以评估大肠杆菌中的其他蛋白质是否可以作用于它。在这些实验中,用大肠杆菌蛋氨酸营养不良菌株B834(DE3)pLysS的粗裂解液对1 mm的TFSeM(as 7b)进行不同时间的孵育,并用LC-MS分析反应。如图10A和B所示,TFSeM和SeM均以时间依赖的方式消耗。当用TFSeM和粗裂解液进行类似反应时煮沸10分钟或在含有分子量为3 kDa的膜的旋转浓缩器中离心的粗裂解液滤液,在整个18小时的培养期内,SeM和TFSeM的浓度保持恒定(图S2A-D)。这种行为强烈地表明,TFSeM在大肠杆菌中是由一种未经鉴定的蛋白质作用的。总之,这些结果表明,尽管TFSeM未转化为TFSeSAM,但它是大肠杆菌中另一种蛋白质的靶点,最有可能将其转化为高硒半胱氨酸或该代谢物的前体。
结论
以N-(叔丁基羰基)-L-天冬氨酸叔丁基酯为原料,经七步反应合成了一种新的非天然氨基酸三氟硒代蛋氨酸(7)。与硒代蛋氨酸(5)相比,三氟硒代蛋氨酸对人结肠癌HCT-116细胞的细胞毒性增强。通过计算和实验验证了这种增强活性的机制解释。用EXAFS和DFT计算5和7的结果表明,它们在光谱和结构上非常相似。然而,当用7(as 7b)和蛋氨酸以9:1的比例在大肠杆菌蛋氨酸营养不良细胞系RF11中表达两个不同的蛋白GB1突变体时,发现令人惊讶的是,超过80%的蛋白含有5,即使没有添加5,这意味着三氟甲基从7失去。一种未知的酶催化这个过程。确定是哪一种酶导致了7的三氟甲基的缺失是很有意义的,因为这种化学基团存在于许多药物中,因此具有内在的药用价值。
本文由福山生物整理翻译,转载请注明出处。