






最新资讯



-
电话: 400-113-6988 邮箱: dongfangxicao@163.com 地址: 深圳市南山区粤海街道高新中三道9号环球数码大厦19楼
↓ 微信扫码识别关注我们 ↓ 
异硒氰酸酯类化合物的合成及生物应用研究进展
发表于:2021-02-20 作者:admin 来源:本站 点击量:22161
原文:Frieben E , Amin S , Sharma A K . Development of Isoselenocyanate Compounds' Syntheses and Biological Applications[J]. Journal of Medicinal Chemistry, 2019.
翻译:
简介
异硫氰酸酯(ISCs)的化学和生物学最近得到了发展,这是由于发现了合成它们的新方法,以及发现了它们对各种疾病,特别是癌症的治疗前景。癌症是全球死亡的主要原因之一,2012年新增病例1410万例,死亡820万例。随着人口的增长和老龄化,以及持续的生活方式行为,如不良的饮食、缺乏体育活动和吸烟,被认为会增加患癌症的风险,这些数字预计会上升。到2030年,预计新病例和死亡人数将分别增至2170万和1300万。因此,癌症是一个重大的健康问题,是社会的重大负担;随着预计的数字增长,迫切需要开发新的、有效的疗法。
硒是一种重要的微量元素,已被证明具有化学预防和化疗活性。事实上,许多流行病学研究表明,硒的摄入与癌症风险呈负相关(参见ref 3)。在三分之二的体内化学和病毒致癌研究中,发现补充硒可降低肿瘤发病率。此外,有机硒化合物已被证明可以抑制化学致癌的起始和起始后阶段。在癌细胞中,硒化合物也表现出抗肿瘤、抗血管生成和促氧化活性。
虽然潜在的抗癌作用机制是复杂的,并没有完全了解,他们被认为与硒化合物的能力有关,能诱导DNA损伤,调节细胞周期,抑制细胞生长,诱导凋亡,并产生活性氧(ROS)。然而,硒化合物的生物学效应是物种形成和浓度依赖的。例如,营养预防癌症(NPC),硒和维生素E预防癌症试验(SELECT)临床试验的相互矛盾的结果证明了物种依赖关系。尽管相同剂量的硒(200微克 /天),发现补充与富硒酵母减少癌症发病率在全国人大的试验中,在补充硒代蛋氨酸(SeMet),硒酵母的主要组成部分,导致没有减少前列腺癌的发病率在选择试验。关于浓度依赖,膳食水平(55微克 /天)的硒化合物是预防各种癌症的发展,而高硒浓度(> 350微克/天)可以致癌,基因毒性和细胞毒性。低剂量硒的化学预防作用通常归因于化合物的能力作为一种抗氧化剂,控制细胞氧化还原状态,防止氧化应激。相反,高剂量使化合物变得氧化还原活性强,对肿瘤细胞有细胞毒性,由于Warburg效应,肿瘤细胞中ROS水平升高,相应的ROS水平降低。此外,与健康对照组相比,患有黑色素瘤、乳腺癌、卵巢癌、结肠癌、头颈癌和胰腺癌的患者全血或血清中的硒水平也有所下降。因此,基于它们的双重化学预防和化疗作用,除了在癌症患者中观察到的硒缺乏外,有机硒化合物具有巨大的致癌潜力,近年来引起了新型硒化合物的合成和开发。特别是,目前最突出的一个例子是ISCs的发展,即天然异硫氰酸酯(ITCs)的硒同位素。

近年来,食品因其可得性、安全性和低成本等特点,作为潜在的化学预防制剂而受到人们的关注。事实上,有很多流行病学研究表明,水果和蔬菜的摄入量与主要场所患癌症的风险呈负相关。特别是,十字花科蔬菜的食用量与癌症风险之间存在很强的负相关关系。十字花科蔬菜,如球芽甘蓝、花椰菜、花椰菜、卷心菜和豆瓣菜,都富含硫甙类化合物。糖苷酸的生物活性分解产物,即ITCs和吲哚,是由液泡水解酶芥子酶的活性产生的。这些具有生物活性的化合物在许多体外和体内研究中已经显示出对不同的人类癌症具有大量的抗癌和化学预防活性。此外,ITCs已被证明与标准化疗协同作用,以增强肿瘤消退。
ITCs的抗癌作用很大程度上归因于其改变解毒途径的能力。ITCs已被证明可以抑制I期细胞色素P-450 (CYP)酶参与前致癌物的激活。此外,他们还诱导了II期酶,可以解毒I期酶产生的剩余亲电代谢物。此外,通过转录刺激抗氧化酶和蛋白质,以及增强自由基清除特性,维生素c可以保护机体免受氧化应激。ITCs也证明了这一点抗肿瘤,抗血管生成和抗转移活性。也就是说,它们已被证明可以抑制细胞周期进展,与巯基结合,产生ROS,并在人类癌症中诱导细胞凋亡。此外,它们还能改变雌激素的代谢,下调雌激素受体,调节丝裂原活化蛋白激酶(MAPK)的信号传导和蛋白激酶C (PKC),抑制组蛋白去乙酰化酶(HDAC),调节细胞核的活化因子k B (NF-κB) NF-E2 p45相关因子2 (Nrf2)和信号传感器和转录激活3 (STAT3),并有选择地耗尽p53突变。然而,在不同p53状态(野生型、突变型、敲除型)的细胞中,研究了ITCs的抗癌活性,表明其抗癌作用是p53依赖还是p53独立尚不清楚。
先前的研究表明,ITCs的活性随着其化学成分的变化而变化。例如,高亲脂性或增加ITCs的烷基链长度已被证明能够增强抑制4-(甲基亚硝胺)-1-(3-吡啶基)-1-丁酮(NNK)诱导的A/J小鼠的肺癌生成。在头颈部鳞癌中,随着ITC烷基链长度的增加,其效价也增加。相比之下,其他结构-活性关系(SAR)研究报道烷基链长度影响但不直接与抑制效力相对应,最佳链长随癌症类型的不同而不同。尽管有这些发现,ITCs已被证明对癌细胞有优先的细胞毒性。特别是异硫氰酸苯乙酯(PEITC)不仅表现出较高的选择性,相对于非恶性卵巢上皮细胞系(T72),其选择性指数(SI)为3.9,但其选择性也高于顺铂(SI = 1.0)。
因此,所建立的化学预防效应使得ITCs成为结构优化的先导化合物。在观察的基础上发现较多硒化合物在癌症预防方面比它们的硫类似物活跃,以及大量的证据支持有机硒化合物用于治疗和预防癌症,非典以异硫氰酸盐为研究对象,用异硫氰酸盐的等容硒代替硫,制备了新型异硒硫氰酸盐。与相应的肿瘤细胞相比,这些新的肿瘤细胞对癌细胞有更强的细胞毒性。鉴于这些非典型肺炎,本研究的目的是探讨ISCs作为潜在的化疗和化学预防药物的用途。特别地,我们总结了近年来在黑素瘤、肺癌、结肠癌、肝癌、前列腺癌和乳腺癌以及其他疾病的合成和发展方面的进展,以及在其他化学领域的应用。

异硒氰酸酯的化学
化学上,ISCs是什么?首先,ISCs是各种有机硒化合物(如杂环化合物、硒代氨基甲酸盐和硒代脲类化合物)的前体和必要中间体,它们都具有生物活性。除了作为具有生物活性的化合物的前体外,isc还被研究和用于许多化学学科的各种应用(例如,,有机,计算,理论,无机,有机金属,和聚合物化学),这将是重点和讨论在下面的小节。
ISCs的制备方法和相关的挑战。
传统上,有机异氰酸酯是通过在氯仿或四氢呋喃(THF)中直接添加元素硒来合成的(图1)。尽管这种方法提供中等到高产量的异氰酸酯,尽管元素硒比其它硒试剂更便宜,该方法的一个缺点是异氰化物的毒性和极刺鼻的气味。此外,合成所需的异氰酸酯起始物质可能需要苛刻、激烈的条件,这可能会改变分子中其他敏感的功能。在三乙胺存在的情况下,使用前体胺、CSe2和HgCl2合成了ISCs(图1)。合成的ISC和胺-氯化汞加合物导致副产物如硒脲、碳二亚胺或异氰化物的形成,这可能使产物的分离和纯化变得复杂。
虽然可以使用其他方法来获得ISCs,但它们的应用范围有限。这些替代方法包括硒化的异氰酸酯反应磷(V)(图1),反应的硒化钠N-arylcarbimidic二氯化(图2),以及光化学重排硒氰酸酯(图1)。ISCs也被一锅法环加成合成,通过腈氧化物与硒代酰胺的反应(图2)。
此外,酰基异氰酸酯直接由酰氯和硒氰酸钾合成(图2)。事实上,三苯基甲基异氰酸酯最初被描述为一种硒氰酸酯,但后来发现是一种异氰酸酯。以同样的方法制备了咪唑基ISCs,并以咪唑基氯作为起始反应物(图2)。
Barton等人开发了一种方便的一锅法制备高产ISCs。本方法以甲苯中相应的甲酰胺为原料制备ISCs;福尔酰胺在硒元素和Et3N等碱基存在的情况下与光气进行脱水(图3)。此后,其他研究人员对该方法进行了改进,以防止中间化合物聚合并提高产量。这样的修改包括了用低危险性的三光气取代了高毒性的光气,以及使用二氯甲烷代替甲苯(图3),作为反应回流温度较低被认为导致更少的聚合。这些修饰已被证明能成功地生成新的苯基烷基异硫氰酸酯、芳基异硫氰酸酯和糖基异硫氰酸酯,且产率较高。该方法的另一个变化涉及到甲酰胺与碳酸二(三氯甲基)和硒的反应(图3)。
近年来,Zakrzewski等人提出了一种合成ISCs的新方法,该方法涉及一种在强碱性条件下操作的双相水/有机体系。特别是,当Se加入到各种异氰化物或相应的胺中(图1),在相转移条件下(50% NaOH, CH2Cl2,铵盐Aliquat 336)可以形成ISCs。上述ISCs合成方法的概述见图1 - 3。
这些研究表明,合成异硫氰酸酯的方法仍在发展中,因为异硫氰酸酯是各种合成异硒氰酸酯的有用原料或中间体硒有机化合物。除了前面提到的ISC合成方法的注意事项外,还可能出现其他有关ISC稳定性的问题。例如,HNCSe在水溶液中不稳定,在气相中不稳定,在低温惰性基体中的宽带紫外线照射下不稳定,迅速分解为氰化氢和硒。其他ISC对阳光很敏感,可以分解释放硒元素。此外,虽然ISC基团已经成功地与过渡金属配合物结合,但ISC基团在与金属配合物结合后,通过失去硒,也被证明可以快速分解。因此,这些稳定性挑战进一步加强了对更新、更简单、更安全的ISC合成方法的需求。
ISCs在化学其他领域的应用。
已知ISCs是含有- NHC(Se) -单位的有机硒化合物的前体,而- NHC(Se) -单位经过脱质子作用后可作为金属的单阴离子Se -配体。因此,在配位化学中对ISC类化合物作为前体化合物进行了研究,可用于制备各种金属配合物的配体。例如,Ben Dahman Andaloussi等人使用更安全的方法从三甲基氯和硒氰酸钾中制备了三甲基ISC。考察了三甲基异硫氰酸酯制备功能化有机硅化合物的反应性和能力;研究发现,三甲基异硫氰酸酯与初级胺反应生成硒脲衍生物,与肼反应生成三甲基硒脲。然后,硒酰脲与水杨醛发生缩合反应,以合理的收率得到亚胺。
除了作为金属配合物配体的前体外,由于ISC阴离子具有很强的配位能力,ISC本身也被用作各种金属配合物中末端或桥联配体的组成部分。虽然ISC基团已成功地与许多过渡金属配合物结合,但也发现ISC基团在与金属配合物结合后,通过失去Se而迅速分解。有趣的是,这种不稳定性只在配位的ISCs中发现,不会自由分解形成氰化物。
郑等人对不同含双(ITC)或双(ISC)配体的铁(II)配合物的热自旋交叉(SCO)行为、过渡温度和SCO的协同性进行了比较。发现有几个ISC类似物与相应的ITC类似物具有相似的结构和分子结构。ISC离子(II)配合物的SCO转变温度高于相应的ITC铁(II)配合物。对于某些ISC配体,在铁(II)配合物晶体结构中的分子间氮-氢-硒键的存在对提高SCO的协同性具有重要作用。
其他的工作已经证明ISC或ITC阴离子加入到含有一维微孔的镍(II)配位聚合物框架中。由于微孔客体分子(如水、甲醇、乙醇等)的羟基可以作为氢键受体与S或Se相互作用,这些配位聚合物具有相似的电负性但不同的范德华半径,因此对其多孔性进行了比较。拉曼光谱分析、红外光谱分析、x射线衍射和热重分析表明,虽然硒和客体分子之间形成了氢键网络,但用硒取代硫对微孔的大小、形状、体积和解吸行为的影响较小在镍(II)配位聚合物的骨架中。
以ISCs为中间体的合成方法也被用来开发新的多组分聚合反应(MCPs),用于合成高级顺序调节聚合物。从这些方法中产生的硒大分子可能在开发用于高级成像的新生物材料或材料方面具有应用价值。最近的工作已经证明了元素硒引入聚合物链。特别是以元素硒为单体,合成了一种新型聚硒脲类聚合物。在MCP反应中,元素Se与烷基异氰酸酯原位反应生成ISCs,再与胺类单体反应生成连接基元。结果表明,该方法在室温和大气条件下反应速度快,原子经济性达到100%。
ISCs的理论和计算研究
ISCs已在许多理论研究中被用作中心焦点或更好地理解其他各种物理现象的手段。例如,研究了异氰酸盐、尿素和含有各种硫(氧(O)、硫(S)或硒)的酰胺的电子离域,以解释C - N旋转势垒和N-反转势垒的趋势。在异氰酸盐中,从头算分子轨道(MO)和密度泛函理论(DFT)的计算表明,C - N的部分双键性质和电子离域性按O < S < Se的顺序增加。ISCs具有最大的部分双键性质和电子离域性。自然键轨道(NBO)分析表明,所观察到的电子离域趋势与硫原子的电负性无关。相反,轨道相互作用形成了观测趋势的基础:参与离域的相互作用轨道之间的能量差越小,共振越强。
除了电子离域之外,我们还利用动态核磁共振波谱和DFT研究了环戊二烯异氰酸酯、异氰酸酯和异碲氰酸酯的重排能力。而五苯基环戊二烯基异氰酸酯和五苯基环戊二烯基ITC仍保留结构刚性, 五苯基环戊二烯基 ISC经历了一个可逆的hetero-Cope重排,产生一个异构硒氰酸酯,硒氰基基团经历了低能量1,5-迁移沿着环戊二烯环的周长变化。异氰酸酯型和ITC型未重排的原因是其热力学稳定性优于相应的异氰酸酯型和硫氰酸酯型。相反,异构ISC和硒氰酸酯能量近似,因为次级轨道之间的相互作用的π-电子环戊二烯环和硒的p-z轨道,它允许发生重排。根据这一发现,五苯基环戊二烯基ISC基团3,3-移位的活化势垒比未取代的ISC低7.6千卡每摩尔,这是由于电子密度在苯基取代形式中有更好的定位。
Trujillo等人使用DFT方法研究了卤素(F、Cl、Br)和甲基取代基对硒氰酸盐和异氰酸酯结合和稳定性的影响。卤代硒氰酸盐比卤代异氰酸盐更稳定,而甲基化异氰酸盐比甲基化硒氰酸盐更稳定。这些观察结果归因于键合行为。即,硒-卤素键(F、Cl、Br)具有较强的离子性,键的离解能比硒与甲基结合时大,而后者具有共价键性,键的离解能较弱。此外,氮-甲基键比氮-卤素(F, Cl,Br)键更强,具有更大的离解能。甲基化形式的CN键保留了三键性质,而卤素衍生物的CN键则减弱并表现出双键性质。此外,随着ISC的卤化,在键临界点(BCP)的电子密度降低,这意味着当从硒氰酸酯异构体转变为ISC时,系统发生了更大的失稳;这一结果与ISC的甲基化形成对比,在CNBCP上可以看到更大的电子密度。
光谱和计算方法也被用来研究振动和旋转,并阐明异硒氰酸酯基团在不同ISCs中的旋转构象。例如,异硒氰酸HNCSe在水溶液中不稳定,迅速分解为氰化氢和硒,因此必须用间接方法测定其理化性质。气体HNCSe的振动和旋转光谱的比较表明,异氰酸不属于刚性、弯曲的分子,而属于准线性化合物。
以甲基异硫氰酸酯为例,其表征在不同的研究中存在差异。富兰克林等人的红外研究表明,甲基ISC的甲基群具有基本的自由内旋。在另一项研究中,发现甲基异硫氰酸酯的微波波谱适合刚性转子,与异氰酸甲酯和ITC的近似相反,后者不符合半刚性转子近似。假设NCSe原子间键的线性关系,计算出CNC键的角度为157°。这一发现与其他测量HNC−X、CNC−X或C2H5−X键角的研究一致,其中键角随着X = O < X = S < X = Se而增加。
相反,对甲基异硫氰酸酯的微波波谱的重新研究表明,甲基异硫氰酸酯与非刚性、准对称的顶分子相一致。计算得到的数控弯曲振动势函数是非调和的,数控键合角为161.3°。对基态能量和线性障碍的解析表明,基态能量高于线性障碍。此外,发现数控弯曲模式的第一振动激发态的能量高于地面振动态。
因为有相对较少的气态ISC文献报道, Møllendal等人合成乙烯基ISC及其MW光谱研究,完成结合量子力学计算。以往的微波研究表明,乙烯基异氰酸酯可以作为共面或反平面的共面,而乙烯基ITC仅作为反平面旋转剂存在。而乙烯ISC一直在预测存在基团或反围平面形式,四种不同的量子计算方法使用Møllendal等人都预测,数控债券的乙烯ISC 反围平面形式存在。通过对数控弯曲振动势函数的计算,发现了极低和非谐极低的平面内弯曲振动以及极多变的离心畸变常量。基于这些观察之前,除了计算数控键角从151到170°(取决于量子化学的计算方法),这些特征研究结果表明,乙烯ISC应该归类为有拟线性CNCSe原子链接而不是刚性的,弯曲反围平面形式。
最近,人们利用紫外光电子能谱和量子化学计算对各种不饱和或伪不饱和isc(乙烯基、2-丙烯基和环丙基isc)的物理化学性质进行了研究,以确定NCSe与不饱和基团之间的相互作用。并对相应的不饱和异氰酸酯和ITC进行了比较。通过对第一电离能的分析和比较,揭示了原子影响IE和最高占有分子轨道(HOMO)能量。即,与乙烯基异氰酸酯相比,乙烯基ITC和乙烯基ISC的IE值降低,HOMO能量增加,说明p-轨道贡献很重要,并起到减弱作用,乙烯基取代基与- NCX (X = S, Se, O)基的相互作用强度,以及降低乙烯基ITC和乙烯基ISC的第一IE。环丙基类似物也有类似的效果。特别是对于异氰酸环丙酯,由于氧原子的作用,其IE值明显高于相应的S或Se类似物。因此,这些结果表明,ITCs和ISCs之间的相互作用较弱,与异氰酸盐相比,其不饱和组分和- NCX结构。此外,在烯丙基不饱和结构中,用S或Se取代- NCX中的氧原子会导致HOMO的建立。只有在异氰酸烯丙酯的情况下,异氰酸酯基团之间的相互作用并观察到烯丙基,作者由此得出结论,必须考虑两个不相互作用的官能团。
异硒氰酸酯在癌症和其他疾病中的应用
除了有许多有用的化学应用外,一些isc也被证明具有强大的生物活性。ISC化合物已被证明对几种疾病类型有效;它们作为抗癌药物的应用已被广泛认可。这些化合物的生物活性将在以下章节中根据它们有效的疾病进行强调。
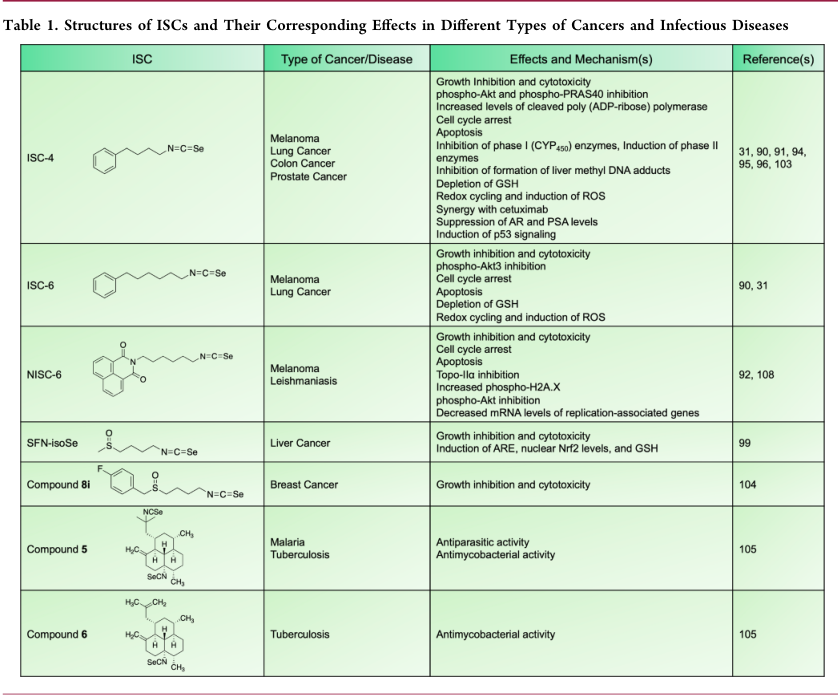
癌症。在过去的十年中,研究已经显示出各种各样的ISCs对癌症的有效预防和治疗作用。体外和体内模型均显示,与相应的ITCs相比,ISCs对癌细胞的细胞毒性更大。此外,这种巨大的治疗潜力已被证明在各种不同的癌症,如黑色素瘤,肺癌,肝癌等等。
黑素瘤。几种不同的isc已被证明对黑色素瘤模型有效。Sharma等人比较了苯基烷基ISCs的生物活性,其中苯基环和ISC片断之间的烷基链长度不同。这些化合物包括苄基异硒氰酸酯(ISC-1)、苯乙基异硒氰酸酯(ISC-2)、苯基-丁基异硒氰酸酯(ISC-4)、苯己基异硒氰酸酯(ISC-6)。在体内和体外的抗癌效果,化合物的稳定性随烷基链长度的增加而增加。具有4-和6-碳链长度的ISC-4和ISC-6被认为是最有效的化合物。因此,Sharma等人后来证明,ISC-4和ISC-6(表1)在降低UACC 903、1205 Lu、WM115黑色素瘤细胞的存活率方面比它们对应的ITCs,即异硫氰酸苯丁酯(PBITC)和异硫氰酸苯己酯(PHITC)更有效。他们也被发现在降低细胞活力方面比苯己基硒酸盐更有效(PHSC)或Akt抑制剂API-2。此外,黑素瘤细胞对ISC-4比正常的人成纤维细胞更敏感,这表明这些化合物可能对癌细胞有选择性。进一步的分析还表明,在体内外,ISC-4和ISC-6抑制Akt3信号,这一信号通路激活了约70%的黑色素瘤,并参与减少黑色素瘤细胞凋亡。这可以通过Akt和Akt底物40kda (phospho-PRAS40)中富含磷脯氨酸的Akt底物的减少得到证明。此外,无论是在体外还是在移植瘤模型中,ISC-4和ISC-6不仅抑制Akt信号通路,而且在低于相应的ITCs的剂量下诱导细胞凋亡。ISC-4后来被选作进一步研究,而不是ISC-6,因为它被认为更像药物,具有良好的LogP值(cLogP 4.4)和分子量(MW = 238),这符合利平斯基关于药物相似性的“五法则”。相比之下,ISC-6亲脂性更强,LogP值较低(cLogP 5.5)。
因此,基于这些发现,我们的小组试图阐明ISC-4对早期黑素细胞病变和正常皮肤细胞的影响。我们证明,与正常黑素细胞相比,ISC-4在诱导来自放射状生长阶段的黑素细胞病变细胞和晚期黑素瘤细胞的细胞毒性方面是2- 5倍。ISC-4可降低Akt3活性,诱导黑色素细胞病变细胞和晚期黑色素瘤细胞凋亡。此外,在没有全身毒性的情况下,在裸鼠身上局部应用ISC-4可使肿瘤体积缩小77%。同样,系统给予ISC-4可忽略器官相关毒性,提示ISC-4可能是治疗黑色素瘤的一种可行的治疗方法,且无明显的脱靶效应。
通过萘酰亚胺基取代苯环生成双拓扑异构体-萘酰亚胺异硒氰酸酯(NISC),进一步优化了苯基烷基异氰酸酯II和Akt通路抑制剂。在米托萘胺的认识基础上,一个萘酰亚胺类似物和强有力的拓扑异构酶-IIα(Topo-IIα)抑制剂, 临床试验失败了,由于系统性毒性相关的问题,我们的研究小组进行了SAR最初修改和了解ITL类似物的功能获得不同烷基链长度NITC类似物。因为ITC被证明可以减少与之相关的全身毒性问题, ITC组进一步取代ISC根是在上述研究的基础上,为了或得一个更安全,双重Topo-IIα/ Akt抑制剂。
在类似物中,具有异硒氰酸酯功能的NISC-6通过6碳烷基链与萘酰亚胺连接(表1),被确定为先导化合物。它在野生型(WT)和BRAF V600E突变型黑素瘤细胞系中同样有效,与其他类似物相比,48和72小时时细胞毒性最强。此外,尽管NISC-6在细胞毒性方面不如米托那非,但与米托那非不同的是,它对癌细胞的选择性超过了正常人类皮肤成纤维细胞(nHDF)。更一步说,硅分子对接,进一步结合体外研究,不仅表明NISC-6融入一种蛋白激酶的活性部位和拓扑酶-IIα,但也表明Topo-IIα活动抑制,以及一种蛋白激酶的磷酸化。此外,NISC-6引起的细胞死亡被发现是由于细胞凋亡的诱导。肿瘤生长在异种移植小鼠模型中,与经药物治疗的小鼠相比,黑素瘤被抑制了约69%,且未观察到明显的全身毒性迹象。因此,肿瘤抑制数据结合体外研究表明,NISC-6是一种可耐受的双作用治疗药物,具有治疗黑色素瘤的潜力。
肺癌。目前,已有两篇关于肺癌中ISCs的报道。在A/J小鼠体内的研究,基于已知的肺癌中ITCs的化学预防特性,评估了ISC-4的化学预防潜力。给药单剂量的ISC-4导致血清、肝脏和肺硒水平随时间的增加而升高,表明ISC-4在小鼠体内具有口服生物利用度。ISC-4通过抑制I期酶和诱导II期酶,调节肝脏和肺组织中I期(cyp450)和II期代谢酶的相对mRNA表达和活性水平。此外,在喂食含有ISC-4的小鼠中,使用烟草特异性前致癌物NNK处理后,观察到肝脏甲基DNA加合物的形成减少和抑制。因此,这些研究表明,ISC-4可能是一种合适的肺癌化学预防剂,因为它具有抑制致癌物代谢和增加解毒的抗启动作用。
随后的体外的SAR结果比较了不同苯基烷基ISCs对增加烷基链长度的作用(ISC-1、ISC-2、ISC-4、ISC-6;图5)在A549肺腺癌细胞中,与相应的苯烷基ITCs。与其他类型癌症的研究相比,ISCs在诱导癌细胞死亡方面比相应的ITCs更有效。Crampsie等人发现,虽然ITCs和ISCs都消耗了还原型谷胱甘肽(GSH),但ISCs比ITCs更快地完成了消耗。相比之下,ITCs的耗竭对GSH的降低作用大于ISCs。此外,与ITC类似物相比,ISCs表现出更强的氧化还原循环能力和诱导ROS水平。有趣的是,只有ITCs能诱导细胞周期阻滞,意味着细胞内的目标在S-和se -取代的类似物之间是不同的。
结肠癌。在几种不同的结肠癌细胞系中,ISC-4以剂量依赖的方式有效地抑制细胞生长。在HT-29结肠癌细胞中,发现ISC-4比其相应的硫类似物(PBITC)以及商业上可用的Akt抑制剂API-2更有效。在结肠癌的异种移植模型中,ISC-4降低了肿瘤的生长速度,但没有明显的全身毒性。肿瘤组织分析显示,ISC-4组小鼠中Akt1、前列腺凋亡反应蛋白-4 (Par-4)和磷酸化akt水平均下调。添加5-氟尿嘧啶(5-FU)治疗标准可增强ISC-4引起的生长抑制。此外,随着肿瘤抑制蛋白Par-4的加入,肿瘤生长进一步减慢。
在另一项研究中,我们评估了ISC-4的协同能力。为了观察ISC-4是否可以改善结肠癌的疗效,我们单独比较了ISC-4在RKO和SW480细胞中的活性,或者将其与19种FDA批准的抗癌疗法联合使用。在这些组合中,发现ISC-4与西妥昔单抗的组合具有协同作用。特别是,这种协同作用只在RKO细胞系中观察到,其中包含野生型KRAS:这一发现与野生型KRAS对西妥昔单抗疗效的要求是一致的。因此,在野生型KRAS细胞系中,ISC-4和西妥昔单抗协同抑制细胞增殖,其作用依赖于ISC-4的剂量,但独立于西妥昔单抗。同样,这种效应与磷酸化akt水平的降低和凋亡的增加有关。此外,在5- FU耐药结肠癌模型中,发现ISC-4和西妥昔单抗的协同作用在体内外均被保留。此外,体内研究表明,药物的组合是可耐受的,不会造成任何明显的毒性。总的来说,这些报道表明ISC-4有可能作为结肠癌治疗药物,既可以作为单一药物,也可以与化疗联合使用。
肝癌。ITC,即萝卜硫素(SFN),是Nrf2/抗氧化反应元件(ARE)信号通路最有效的诱导因子之一。在完全发展的癌症中,然而,Nrf2激活是一把双刃剑,因为Nrf2激活调节参与抗癌药物解毒的通路,可能导致癌症细胞耐药。sfn介导的这一途径导致GSH生物合成酶如限速谷氨酸半胱氨酸连接酶(GCLc)和II期解毒酶的上调。因为增强谷胱甘肽及其感应SFN的化学预防,扮演着重要的角色,由于已知化学预防 Se化合物的影响等, 假设ITC部分的硫原子与硒的替代可能使硫代嘌呤异硒氰酸酯(SFN-isoSe)(表1)具有更强的诱导ARE信号和Nrf2的能力。这一假设首先在HepG2中进行了6小时的药物治疗。而生存能力并不影响复合剂在或低于10μM,观察对细胞生存能力减少50%在SNF-isoSe的浓度为20μM时。结论是在6小时的药物暴露后,ISCs比ITCs更有效地降低细胞活力。也就是说,HepG2细胞的活力降低到对照组的0、14和0%,分别对应于sfn -异硒、苯乙基异硒氰酸酯(PEISC,或ISC-2)和苯乙基异硒氰酸酯(BISC,或ISC-1)。
类似于可行性研究,ARE诱导效应是相同的在SFN和SFN-isoSe之间,当剂量为0,5和10微摩尔时。在20μM剂量时, 由SFN-isoSe诱导荧光素酶活动水平是SFN的两倍多。与对照化合物PEISC和BISC不同,对于SFN-isoSe, ARE的增加和细胞活力的降低是剂量依赖性的。
在非癌小鼠胚胎成纤维细胞(MEF)中,无论浓度如何,SFN- isoSe诱导的核Nrf2水平也高于SFN。在MEFs中进一步的实验表明,SFN-isoSe诱导GSH的能力是依赖于Nrf2,与SFN相反,SFN显示谷胱甘肽耗竭。综上所述,这些发现表明,SFN-isoSe提高核Nrf2导致GSH酶(如GCLc)的上调,最终导致GSH水平升高。SFN- isoSe诱导GSH和SFN消耗GSH也表明,SFN- isoSe对MEFs的毒性小于SFN。因此,这些结果,与SFN- isose比SFN赋予癌细胞更大的细胞毒性相结合,表明SFN- isoSe可能是一种潜在的化学预防剂。
前列腺癌。Wu等人之前已经证明了ISC-4对其他类型癌症的疗效,他们研究了ISC-4对前列腺癌的影响。ISC-4在降低LNCaP细胞活力和诱导方面的作用是相应S类似物PBITC的4倍。与早期报道的结肠癌和黑色素瘤的研究结果相反,ISC-4并没有抑制Akt的磷酸化,这说明在LNCaP前列腺癌细胞的细胞毒性机制中,Akt的抑制并不是导致ISC-4磷酸化的原因。相反,ISC-4在转录后可降低雄激素受体(AR)和前列腺特异性抗原(PSA)水平,与细胞凋亡无关。
其他实验表明,ISC-4诱导ROS,通过激活p53信号,不仅抑制AR/PSA,而且促进细胞凋亡。特别是p53上调凋亡调节剂(PUMA)-Bax (bcl -2相关的X蛋白)的固有凋亡级联反应被激活。p53的缺失导致了凋亡的衰减,这一效应与ISC-4对AR/PSA的影响以及p53下游靶点的下调无关,也能减弱isc -4介导的细胞凋亡。
乳腺癌。Cierpia?等人合成一系列有机氟ISC萝卜硫素类似物与不同的化学成分,并比较他们的抗癌活动相对于ITC萝卜硫素。由于SFN已知可诱导乳腺癌细胞周期阻滞和细胞死亡,因此在不同的乳腺癌细胞系中筛选了类似物。通过几项测试,发现新的类似物相对于SFN,对癌细胞生长有更强的选择性抑制作用。
在各种类似物中,4-异氰基-1-丁基4 ' -氟代酶亚砜(化合物8i)(表1)被认为是潜在的先导化合物。尽管它在两种被测试的乳腺癌细胞系(MDA-MB-231和MCF7)中都有效力,但它对与激素无关、三阴性的MDA-MB-231细胞选择性最强。MDA-MB-231细胞是一种侵袭性乳腺癌的模型,对传统的、靶向的或基于激素的疗法无效。此外,化合物8i对癌细胞总体上具有选择性:在正常细胞系中,化合物8i的毒性比SFN小,在癌细胞中,化合物8i的细胞毒性比SFN更强。
传染病疟疾。Nieves等人合成了几种基于(−)-8,15-二异氰酸酯-11(20)-亲和素的结构的化合物,亲和素是一种已知的具有有效的抗疟疾和抗细菌作用的代谢物,从海洋海绵Svenzea flava中分离得到。之前SAR研究表明,异氰酸酯的功能是生物活性所必需的,其位置对活性也很重要。记住这一点,根据观察海绵状异氰酸酯-、ITC-和甲酰胺-二萜类化合物也具有生物活性,其新型的二萜类化合物在不同的位置加入了ISCs和ITCs作为潜在的药效基团来测试。在两个不同的恶性疟原虫疟原虫系(氯喹-)中类似物的筛选表明敏感的3D7和耐药的Dd2)显示所有的ISC类似物都表现出亚微摩尔的抗疟原虫活性。这些混合的化合物,化合物5(表1),其中包含两个ISC,比标准更强有力的药物氯喹Dd2 (IC 50 0.0066μM相比0.0519μM)。此外,它对耐药株表现出更大的选择性,与氯喹(SI =4518)相比,具有更高的选择性指数值(SI = 7356)。
肺结核。在上一节所述的同一研究中,Nieves等人也筛选了他们的ITC-和ISC-amphilectane二萜来对抗Mtb h37 Rv,以阐明最低抑制浓度(MIC)。经isc功能化的杂化化合物MIC值最低;特别是,混合化合物5和6的最低抑制浓度为 3.9和2.1μM。相比之下,ITC-功能化的 amphilectane二萜有更低的MIC,从26.8到99.1μM。此外,在杂种化合物5和6中,化合物6被认为是最有效、最具选择性的,这一发现,与ITC -杂交类似物的数据相结合,再次表明硒在这些化合物的选择性和强大的细胞毒性中发挥作用。
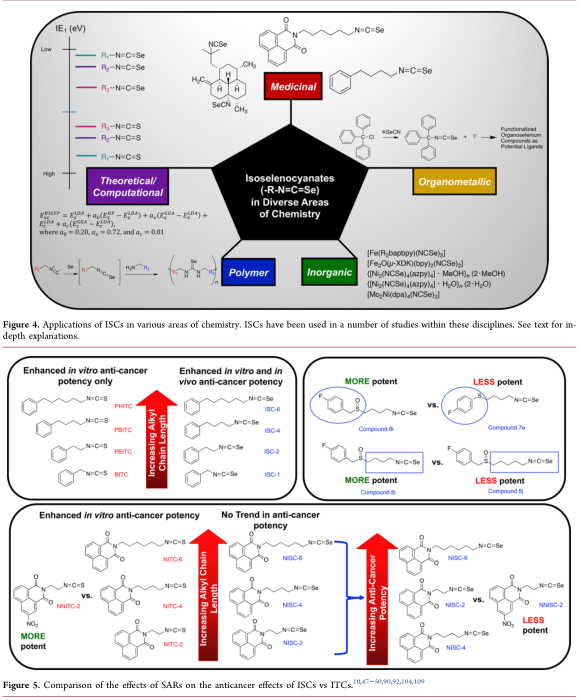
利什曼病。由于硒化合物和抗肿瘤药物均表现出利什曼杀菌活性,因此,我们研究了ISC衍生物在大利什曼原虫和亚马逊利什曼原虫原体和胞内无利什曼原虫形态中的抗增殖活性。在检测的类似物中,发现NISC-6具有抑制利什曼原虫原核细胞增殖的作用,其纳米范围的IC 50值是参考药物两性霉素B的3倍(两性B)。此外,NISC-6的选择性指数最高(SI = 416.7),几乎是参考药物的10倍。虽然L. amazonensis对Ampho B和NISC-6的敏感性较低,但NISC-6的SI (SI = 41.7)仍然是Ampho B的2倍(SI = 20.5)。NISC-6也被证明能引起细胞周期阻滞,证据是复制相关基因如增殖细胞核抗原(PCNA)、拓扑异构酶-2 (TOP2)和小染色体维持复合体4 (MCM4)的mRNA水平下降。此外,与未处理的细胞相比,NISC-6处理导致G1期寄生细胞数量增加,S期减少(表1)。
结论和观点
从前面几节可以明显看出,ISCs在一些化学学科中显示出了很大的应用潜力,如理论、计算、有机金属、聚合物和无机化学(图4)。
然而,在这些用途中,由于缺乏有效的、长期的治疗以及预期的癌症病例和死亡的增加,作为抗癌剂的应用尤其令人感兴趣。此外,所述的研究表明,ISCs是有效和有效的抗癌和化学预防剂,在大多数情况下比相应的ITCs更有效。
通过增加与苯基烷基异氰酸酯(ISC-1、ISC-2、ISC-4、ISC-6)的烷基链长度,桥接苯基环和ISC片断,增加了体外和体内的生长抑制力(图5)。这一发现与以往各种苯基烷基ITCs的体外研究结果相似,即随着烷基链长度增加至8个碳,其效价增加,但与体内研究不同,在体内研究中,烷基链长度增加并不对应体内更强的肿瘤抑制作用。重要的是,在isc研究中,观察到对照硒代半胱氨酸缺乏疗效,这表明单硒并不是iscs增强生长抑制作用的原因,而是isc结构与硒的结合。还推断,较长的烷基链长度可能有助于ISC或ITC基团与酶活性所需的关键残基的接触和结合。
然而,在NISC类似物的情况下,增加的烷基链长度并不总是对应更强的生物活性,尽管确定NISC-6是与6碳烷基链(测试中最长的烷基链)最有效的化合物。这与相应的硫类似物形成对比,硫类似物中增加的烷基链长度增加了效价。此外,不像硫类似物NNITC-2,其中硝基取代萘酰亚胺环系与未被取代的nnc -2相比,NNISC-2的细胞毒性较低,表明在Se类似物的情况下,硝基取代是不利的(图5)。
对于SFN的有机氟ISC类似物,硫原子的氧化,当与4-氟苯环和亚砜基团之间的亚甲基基团结合时,发现增加了抗癌效能。此外,增加硫基官能团和ISC片断之间的烷基链长度(从4个碳到5个碳)会导致对正常细胞系的细胞毒性增加,从而降低化合物的癌细胞选择性(图5)。
根据对这些SARs的观察,可以得出结论:氧化态、环取代和烷基链长度对ISCs的效能和效能有影响。同样,这些因素都有助于这些化合物的药物相似性,这是一个必须考虑的性质,以优化和提高这些化合物作为潜在的抗癌疗法和化学预防剂。因此,在上述的一些SAR研究中已经考虑了药物相似性。例如,计算分析表明,NISC-6符合Lipinski’s Rule 5的原则,其计算的拓扑极性表面积预测具有较高的口服生物利用度。此外,使用苯基烷基ISCs, Sharma等人注意到在苯环和ISC之间增加烷基链的长度的功能增加了ITCs和ISCs的亲脂性和CLogP值,但对ISCs的亲脂性和CLogP值的影响更大。随着烷基链长度的增加,化合物应能更容易、更有效地扩散到细胞内。虽然ISC-4和ISC-6同样有效,但只有ISC-4的CLogP符合Lipinski的五项规则。因此,这一知识,以及已知的诱导食道癌变的大鼠由于PHITC的高亲脂性使ISC-6成为不太适合的抗癌和/或化学预防剂。
因此,这些观点又回到了另一个重要的问题:为什么要使用硒?硫和硒是元素周期表上同一家族的两个成员,它们的氧化态相同,这两种元素之间有什么不同之处,使得它们具有更强的抗癌功效和效力?SARs表明,在体内外,ISCs比ITCs具有更强的活性,并具有更强的亲脂性。也许这些生物活动的差异可以根据它们的原子和电子特性而不是仅仅根据它们的分子特性来解释。虽然已知S和Se具有相似的电负性值,但它们的原子、离子和范德华半径以及它们的氢键能力不同。与S相比,Se具有更大的原子、离子和范德瓦尔斯半径,Se原子上的电荷更分散,这削弱了分子间的相互作用。硒有一个比硫更强的π-bonding,此外,虽然S有时被认为是氢键的受体,与Se的氢键可以忽略不计。也许,当整个分子与相应的靶蛋白相互作用时,这两种元素之间的细微差别组合在一起,就可以解释所观察到的抗癌功效和效力的差异。
如前所述,Crampsie等人试图从生物学机制上阐明苯烷基ITCs与ISCs中S和Se的区别。拟一阶动力学实验表明,无论是ITCs还是ISCs,与GSH的反应速率均随烷基链长度的增加而降低。虽然ISCs的反应速度比ITCs快,但在S和Se类似物之间,GSH的平衡浓度没有显著差异。在肺癌细胞中,GSH在ISCs中消耗得更快,但在ITCs中,总消耗更大。也许ISCs更强的反应性可以归因于Se的价电子比硫更不稳定,因为它的C=Se键不如C=S稳定,使得它对巯基亲核试剂的反应更强烈。
与报道的ITCs相似,ISC化合物与亲核试剂的反应活性表明它们在体内系统的整体稳定性较差,从而影响了它们的类药物特性。由于ITC功能的亲电性,天然ITC SFN的半衰期仅为1.77±0.13 h。然而,由于硫醇共轭代谢物ISC和ITC的化合物,与蛋白质反应形成硫醇(如谷胱甘肽、半胱氨酸和防治作用)或在硫醇尿酸形成通路,也被证明是有效的抗癌药物,这些化合物可以一起持续比较久活性代谢物,从而导致异硫氰酸酯和ISC的功效。然而,可以通过创造更稳定的水溶性前药来克服稳定性差的限制,如Jiang等人最近报道的,SFN将在未来创造临床相关的ISC候选药物。
此外,ISCs的氧化还原循环生成ROS的效率更高,范围也更广,这与−SeH的氧化还原循环和反应活性高于−SH的事实一致。因此,这些数据表明,ISCs和ITCs的细胞毒性作用与巯基相互作用有关。尤其是isc,增强细胞毒性效应被认为是由于谷胱甘肽耗竭和氧化还原循环,而与谷胱甘肽反应的速率无关,由于数据反驳了原先的假设,即用Se取代S会降低碳原子的亲电性,从而降低硫醇的反应活性。
无论如何,isc已显示出作为潜在癌症治疗或化学预防剂的巨大潜力。除了单纯的ISC-4和西妥昔单抗外,未来癌症相关的ISC研究应该寻找其他潜在的联合治疗方法。例如,可以双重Akt Topo-IIα抑制剂与BRAF V600E抑制剂PLX4032 NISC-6协同作用,帮助延缓其在黑色素瘤中的耐药性?当有机氟ISC萝卜硫素类似物与标准化疗联合使用时,三阴性乳腺癌的侵袭性是否可以降低?
此外,所述iscs可以类似于n isc-6的方式进行修饰,其中isc部分和必需基团与已知抑制剂和化疗剂的生物活性功能结合,并且可以将所述修饰后的化合物的生物活性与其各自的母体化合物进行比较。此外,这些iscs的反应性可在并入几个isc和/或itc部分时进行研究。此外,胶质母细胞瘤是一种侵袭性癌症,没有有效的isc类似物。未来的研究还可以集中在合成能够跨越血脑屏障的有效、靶向ISC类似物上。
正如生物学研究所提示的,要获得一种非常有效的、具有癌症细胞选择性和生物可利用性的ISC,需要在优化亲脂性(即、氧化态和/或芳香环取代物而不丧失已知的生物活性。为了获得这些优化的ISCs,还必须有可靠、简单、安全的合成方法。虽然所述的一些合成方法有其局限性,但未来的研究可以努力实现更绿色的方法,以产生所需的isc,并获得良好的产量。这种合成方法的发展将加速这些化合物的结构优化。
除了抗癌的ISCs外,强调ISCs在其他疾病中的作用也很重要。Nieves等人指出,新的ISCs正在作为抗疟疾和抗结核药物开发。通过这些发现的扩展,未来的研究可以集中于开发用于其他传染病的ISCs,如霍乱或脑膜炎。在现有抗菌药物中添加ISC基团,抗菌药物的生物活性部分可与现有的ISC化合物融合,与多种抗感染药物联合使用可评估ISC的协同作用。
作为一个总结,ISCs有能力在未来几年形成许多化学领域,如理论、有机金属、聚合物和无机化学,这已经被他们的研究证明了。它们在药物化学领域以及在癌症和人类疾病方面尤其有前途,因为它们已被证明比同构硫类似物更有效。随着更多的研究和结构优化,这是一个很大的希望,新的ISC化合物以及一些在这篇综述中描述的ISC化合物可以推进到临床试验,并最终在临床作为新的癌症药物和化学预防剂。
翻译:
异硒氰酸酯类化合物的合成及生物应用研究进展
摘要:随着癌症病例和癌症相关死亡人数在未来几年的上升,寻找预防或治疗癌症的方法已迫在眉睫。因此,食品衍生产品因其可得性、安全性和低成本而作为潜在的化学预防剂而受到关注。十字花科蔬菜中含硫硫甙的分解产物异硫氰酸酯对不同的人类癌症具有潜在的抗癌和化学预防作用。此外,已知有机硒化合物具有化学预防和化疗活性;此外,这些化合物是比其硫同位素更有效的抗癌剂。因此,异硫氰酸酯被修饰成异硒氰酸酯,与相应的硫类似物相比,异硒氰酸酯对癌细胞的细胞毒性更强。在此,综述了异硒氰酸酯作为新型癌症和其他疾病治疗药物的合成和发展,重点介绍了该类化合物在化学和计算方面的多样化研究及其相关的生物学应用。简介
异硫氰酸酯(ISCs)的化学和生物学最近得到了发展,这是由于发现了合成它们的新方法,以及发现了它们对各种疾病,特别是癌症的治疗前景。癌症是全球死亡的主要原因之一,2012年新增病例1410万例,死亡820万例。随着人口的增长和老龄化,以及持续的生活方式行为,如不良的饮食、缺乏体育活动和吸烟,被认为会增加患癌症的风险,这些数字预计会上升。到2030年,预计新病例和死亡人数将分别增至2170万和1300万。因此,癌症是一个重大的健康问题,是社会的重大负担;随着预计的数字增长,迫切需要开发新的、有效的疗法。
硒是一种重要的微量元素,已被证明具有化学预防和化疗活性。事实上,许多流行病学研究表明,硒的摄入与癌症风险呈负相关(参见ref 3)。在三分之二的体内化学和病毒致癌研究中,发现补充硒可降低肿瘤发病率。此外,有机硒化合物已被证明可以抑制化学致癌的起始和起始后阶段。在癌细胞中,硒化合物也表现出抗肿瘤、抗血管生成和促氧化活性。
虽然潜在的抗癌作用机制是复杂的,并没有完全了解,他们被认为与硒化合物的能力有关,能诱导DNA损伤,调节细胞周期,抑制细胞生长,诱导凋亡,并产生活性氧(ROS)。然而,硒化合物的生物学效应是物种形成和浓度依赖的。例如,营养预防癌症(NPC),硒和维生素E预防癌症试验(SELECT)临床试验的相互矛盾的结果证明了物种依赖关系。尽管相同剂量的硒(200微克 /天),发现补充与富硒酵母减少癌症发病率在全国人大的试验中,在补充硒代蛋氨酸(SeMet),硒酵母的主要组成部分,导致没有减少前列腺癌的发病率在选择试验。关于浓度依赖,膳食水平(55微克 /天)的硒化合物是预防各种癌症的发展,而高硒浓度(> 350微克/天)可以致癌,基因毒性和细胞毒性。低剂量硒的化学预防作用通常归因于化合物的能力作为一种抗氧化剂,控制细胞氧化还原状态,防止氧化应激。相反,高剂量使化合物变得氧化还原活性强,对肿瘤细胞有细胞毒性,由于Warburg效应,肿瘤细胞中ROS水平升高,相应的ROS水平降低。此外,与健康对照组相比,患有黑色素瘤、乳腺癌、卵巢癌、结肠癌、头颈癌和胰腺癌的患者全血或血清中的硒水平也有所下降。因此,基于它们的双重化学预防和化疗作用,除了在癌症患者中观察到的硒缺乏外,有机硒化合物具有巨大的致癌潜力,近年来引起了新型硒化合物的合成和开发。特别是,目前最突出的一个例子是ISCs的发展,即天然异硫氰酸酯(ITCs)的硒同位素。
近年来,食品因其可得性、安全性和低成本等特点,作为潜在的化学预防制剂而受到人们的关注。事实上,有很多流行病学研究表明,水果和蔬菜的摄入量与主要场所患癌症的风险呈负相关。特别是,十字花科蔬菜的食用量与癌症风险之间存在很强的负相关关系。十字花科蔬菜,如球芽甘蓝、花椰菜、花椰菜、卷心菜和豆瓣菜,都富含硫甙类化合物。糖苷酸的生物活性分解产物,即ITCs和吲哚,是由液泡水解酶芥子酶的活性产生的。这些具有生物活性的化合物在许多体外和体内研究中已经显示出对不同的人类癌症具有大量的抗癌和化学预防活性。此外,ITCs已被证明与标准化疗协同作用,以增强肿瘤消退。
ITCs的抗癌作用很大程度上归因于其改变解毒途径的能力。ITCs已被证明可以抑制I期细胞色素P-450 (CYP)酶参与前致癌物的激活。此外,他们还诱导了II期酶,可以解毒I期酶产生的剩余亲电代谢物。此外,通过转录刺激抗氧化酶和蛋白质,以及增强自由基清除特性,维生素c可以保护机体免受氧化应激。ITCs也证明了这一点抗肿瘤,抗血管生成和抗转移活性。也就是说,它们已被证明可以抑制细胞周期进展,与巯基结合,产生ROS,并在人类癌症中诱导细胞凋亡。此外,它们还能改变雌激素的代谢,下调雌激素受体,调节丝裂原活化蛋白激酶(MAPK)的信号传导和蛋白激酶C (PKC),抑制组蛋白去乙酰化酶(HDAC),调节细胞核的活化因子k B (NF-κB) NF-E2 p45相关因子2 (Nrf2)和信号传感器和转录激活3 (STAT3),并有选择地耗尽p53突变。然而,在不同p53状态(野生型、突变型、敲除型)的细胞中,研究了ITCs的抗癌活性,表明其抗癌作用是p53依赖还是p53独立尚不清楚。
先前的研究表明,ITCs的活性随着其化学成分的变化而变化。例如,高亲脂性或增加ITCs的烷基链长度已被证明能够增强抑制4-(甲基亚硝胺)-1-(3-吡啶基)-1-丁酮(NNK)诱导的A/J小鼠的肺癌生成。在头颈部鳞癌中,随着ITC烷基链长度的增加,其效价也增加。相比之下,其他结构-活性关系(SAR)研究报道烷基链长度影响但不直接与抑制效力相对应,最佳链长随癌症类型的不同而不同。尽管有这些发现,ITCs已被证明对癌细胞有优先的细胞毒性。特别是异硫氰酸苯乙酯(PEITC)不仅表现出较高的选择性,相对于非恶性卵巢上皮细胞系(T72),其选择性指数(SI)为3.9,但其选择性也高于顺铂(SI = 1.0)。
因此,所建立的化学预防效应使得ITCs成为结构优化的先导化合物。在观察的基础上发现较多硒化合物在癌症预防方面比它们的硫类似物活跃,以及大量的证据支持有机硒化合物用于治疗和预防癌症,非典以异硫氰酸盐为研究对象,用异硫氰酸盐的等容硒代替硫,制备了新型异硒硫氰酸盐。与相应的肿瘤细胞相比,这些新的肿瘤细胞对癌细胞有更强的细胞毒性。鉴于这些非典型肺炎,本研究的目的是探讨ISCs作为潜在的化疗和化学预防药物的用途。特别地,我们总结了近年来在黑素瘤、肺癌、结肠癌、肝癌、前列腺癌和乳腺癌以及其他疾病的合成和发展方面的进展,以及在其他化学领域的应用。
异硒氰酸酯的化学
化学上,ISCs是什么?首先,ISCs是各种有机硒化合物(如杂环化合物、硒代氨基甲酸盐和硒代脲类化合物)的前体和必要中间体,它们都具有生物活性。除了作为具有生物活性的化合物的前体外,isc还被研究和用于许多化学学科的各种应用(例如,,有机,计算,理论,无机,有机金属,和聚合物化学),这将是重点和讨论在下面的小节。
ISCs的制备方法和相关的挑战。
传统上,有机异氰酸酯是通过在氯仿或四氢呋喃(THF)中直接添加元素硒来合成的(图1)。尽管这种方法提供中等到高产量的异氰酸酯,尽管元素硒比其它硒试剂更便宜,该方法的一个缺点是异氰化物的毒性和极刺鼻的气味。此外,合成所需的异氰酸酯起始物质可能需要苛刻、激烈的条件,这可能会改变分子中其他敏感的功能。在三乙胺存在的情况下,使用前体胺、CSe2和HgCl2合成了ISCs(图1)。合成的ISC和胺-氯化汞加合物导致副产物如硒脲、碳二亚胺或异氰化物的形成,这可能使产物的分离和纯化变得复杂。
虽然可以使用其他方法来获得ISCs,但它们的应用范围有限。这些替代方法包括硒化的异氰酸酯反应磷(V)(图1),反应的硒化钠N-arylcarbimidic二氯化(图2),以及光化学重排硒氰酸酯(图1)。ISCs也被一锅法环加成合成,通过腈氧化物与硒代酰胺的反应(图2)。
此外,酰基异氰酸酯直接由酰氯和硒氰酸钾合成(图2)。事实上,三苯基甲基异氰酸酯最初被描述为一种硒氰酸酯,但后来发现是一种异氰酸酯。以同样的方法制备了咪唑基ISCs,并以咪唑基氯作为起始反应物(图2)。
Barton等人开发了一种方便的一锅法制备高产ISCs。本方法以甲苯中相应的甲酰胺为原料制备ISCs;福尔酰胺在硒元素和Et3N等碱基存在的情况下与光气进行脱水(图3)。此后,其他研究人员对该方法进行了改进,以防止中间化合物聚合并提高产量。这样的修改包括了用低危险性的三光气取代了高毒性的光气,以及使用二氯甲烷代替甲苯(图3),作为反应回流温度较低被认为导致更少的聚合。这些修饰已被证明能成功地生成新的苯基烷基异硫氰酸酯、芳基异硫氰酸酯和糖基异硫氰酸酯,且产率较高。该方法的另一个变化涉及到甲酰胺与碳酸二(三氯甲基)和硒的反应(图3)。
近年来,Zakrzewski等人提出了一种合成ISCs的新方法,该方法涉及一种在强碱性条件下操作的双相水/有机体系。特别是,当Se加入到各种异氰化物或相应的胺中(图1),在相转移条件下(50% NaOH, CH2Cl2,铵盐Aliquat 336)可以形成ISCs。上述ISCs合成方法的概述见图1 - 3。
这些研究表明,合成异硫氰酸酯的方法仍在发展中,因为异硫氰酸酯是各种合成异硒氰酸酯的有用原料或中间体硒有机化合物。除了前面提到的ISC合成方法的注意事项外,还可能出现其他有关ISC稳定性的问题。例如,HNCSe在水溶液中不稳定,在气相中不稳定,在低温惰性基体中的宽带紫外线照射下不稳定,迅速分解为氰化氢和硒。其他ISC对阳光很敏感,可以分解释放硒元素。此外,虽然ISC基团已经成功地与过渡金属配合物结合,但ISC基团在与金属配合物结合后,通过失去硒,也被证明可以快速分解。因此,这些稳定性挑战进一步加强了对更新、更简单、更安全的ISC合成方法的需求。
ISCs在化学其他领域的应用。
已知ISCs是含有- NHC(Se) -单位的有机硒化合物的前体,而- NHC(Se) -单位经过脱质子作用后可作为金属的单阴离子Se -配体。因此,在配位化学中对ISC类化合物作为前体化合物进行了研究,可用于制备各种金属配合物的配体。例如,Ben Dahman Andaloussi等人使用更安全的方法从三甲基氯和硒氰酸钾中制备了三甲基ISC。考察了三甲基异硫氰酸酯制备功能化有机硅化合物的反应性和能力;研究发现,三甲基异硫氰酸酯与初级胺反应生成硒脲衍生物,与肼反应生成三甲基硒脲。然后,硒酰脲与水杨醛发生缩合反应,以合理的收率得到亚胺。
除了作为金属配合物配体的前体外,由于ISC阴离子具有很强的配位能力,ISC本身也被用作各种金属配合物中末端或桥联配体的组成部分。虽然ISC基团已成功地与许多过渡金属配合物结合,但也发现ISC基团在与金属配合物结合后,通过失去Se而迅速分解。有趣的是,这种不稳定性只在配位的ISCs中发现,不会自由分解形成氰化物。
郑等人对不同含双(ITC)或双(ISC)配体的铁(II)配合物的热自旋交叉(SCO)行为、过渡温度和SCO的协同性进行了比较。发现有几个ISC类似物与相应的ITC类似物具有相似的结构和分子结构。ISC离子(II)配合物的SCO转变温度高于相应的ITC铁(II)配合物。对于某些ISC配体,在铁(II)配合物晶体结构中的分子间氮-氢-硒键的存在对提高SCO的协同性具有重要作用。
其他的工作已经证明ISC或ITC阴离子加入到含有一维微孔的镍(II)配位聚合物框架中。由于微孔客体分子(如水、甲醇、乙醇等)的羟基可以作为氢键受体与S或Se相互作用,这些配位聚合物具有相似的电负性但不同的范德华半径,因此对其多孔性进行了比较。拉曼光谱分析、红外光谱分析、x射线衍射和热重分析表明,虽然硒和客体分子之间形成了氢键网络,但用硒取代硫对微孔的大小、形状、体积和解吸行为的影响较小在镍(II)配位聚合物的骨架中。
以ISCs为中间体的合成方法也被用来开发新的多组分聚合反应(MCPs),用于合成高级顺序调节聚合物。从这些方法中产生的硒大分子可能在开发用于高级成像的新生物材料或材料方面具有应用价值。最近的工作已经证明了元素硒引入聚合物链。特别是以元素硒为单体,合成了一种新型聚硒脲类聚合物。在MCP反应中,元素Se与烷基异氰酸酯原位反应生成ISCs,再与胺类单体反应生成连接基元。结果表明,该方法在室温和大气条件下反应速度快,原子经济性达到100%。
ISCs的理论和计算研究
ISCs已在许多理论研究中被用作中心焦点或更好地理解其他各种物理现象的手段。例如,研究了异氰酸盐、尿素和含有各种硫(氧(O)、硫(S)或硒)的酰胺的电子离域,以解释C - N旋转势垒和N-反转势垒的趋势。在异氰酸盐中,从头算分子轨道(MO)和密度泛函理论(DFT)的计算表明,C - N的部分双键性质和电子离域性按O < S < Se的顺序增加。ISCs具有最大的部分双键性质和电子离域性。自然键轨道(NBO)分析表明,所观察到的电子离域趋势与硫原子的电负性无关。相反,轨道相互作用形成了观测趋势的基础:参与离域的相互作用轨道之间的能量差越小,共振越强。
除了电子离域之外,我们还利用动态核磁共振波谱和DFT研究了环戊二烯异氰酸酯、异氰酸酯和异碲氰酸酯的重排能力。而五苯基环戊二烯基异氰酸酯和五苯基环戊二烯基ITC仍保留结构刚性, 五苯基环戊二烯基 ISC经历了一个可逆的hetero-Cope重排,产生一个异构硒氰酸酯,硒氰基基团经历了低能量1,5-迁移沿着环戊二烯环的周长变化。异氰酸酯型和ITC型未重排的原因是其热力学稳定性优于相应的异氰酸酯型和硫氰酸酯型。相反,异构ISC和硒氰酸酯能量近似,因为次级轨道之间的相互作用的π-电子环戊二烯环和硒的p-z轨道,它允许发生重排。根据这一发现,五苯基环戊二烯基ISC基团3,3-移位的活化势垒比未取代的ISC低7.6千卡每摩尔,这是由于电子密度在苯基取代形式中有更好的定位。
Trujillo等人使用DFT方法研究了卤素(F、Cl、Br)和甲基取代基对硒氰酸盐和异氰酸酯结合和稳定性的影响。卤代硒氰酸盐比卤代异氰酸盐更稳定,而甲基化异氰酸盐比甲基化硒氰酸盐更稳定。这些观察结果归因于键合行为。即,硒-卤素键(F、Cl、Br)具有较强的离子性,键的离解能比硒与甲基结合时大,而后者具有共价键性,键的离解能较弱。此外,氮-甲基键比氮-卤素(F, Cl,Br)键更强,具有更大的离解能。甲基化形式的CN键保留了三键性质,而卤素衍生物的CN键则减弱并表现出双键性质。此外,随着ISC的卤化,在键临界点(BCP)的电子密度降低,这意味着当从硒氰酸酯异构体转变为ISC时,系统发生了更大的失稳;这一结果与ISC的甲基化形成对比,在CNBCP上可以看到更大的电子密度。
光谱和计算方法也被用来研究振动和旋转,并阐明异硒氰酸酯基团在不同ISCs中的旋转构象。例如,异硒氰酸HNCSe在水溶液中不稳定,迅速分解为氰化氢和硒,因此必须用间接方法测定其理化性质。气体HNCSe的振动和旋转光谱的比较表明,异氰酸不属于刚性、弯曲的分子,而属于准线性化合物。
以甲基异硫氰酸酯为例,其表征在不同的研究中存在差异。富兰克林等人的红外研究表明,甲基ISC的甲基群具有基本的自由内旋。在另一项研究中,发现甲基异硫氰酸酯的微波波谱适合刚性转子,与异氰酸甲酯和ITC的近似相反,后者不符合半刚性转子近似。假设NCSe原子间键的线性关系,计算出CNC键的角度为157°。这一发现与其他测量HNC−X、CNC−X或C2H5−X键角的研究一致,其中键角随着X = O < X = S < X = Se而增加。
相反,对甲基异硫氰酸酯的微波波谱的重新研究表明,甲基异硫氰酸酯与非刚性、准对称的顶分子相一致。计算得到的数控弯曲振动势函数是非调和的,数控键合角为161.3°。对基态能量和线性障碍的解析表明,基态能量高于线性障碍。此外,发现数控弯曲模式的第一振动激发态的能量高于地面振动态。
因为有相对较少的气态ISC文献报道, Møllendal等人合成乙烯基ISC及其MW光谱研究,完成结合量子力学计算。以往的微波研究表明,乙烯基异氰酸酯可以作为共面或反平面的共面,而乙烯基ITC仅作为反平面旋转剂存在。而乙烯ISC一直在预测存在基团或反围平面形式,四种不同的量子计算方法使用Møllendal等人都预测,数控债券的乙烯ISC 反围平面形式存在。通过对数控弯曲振动势函数的计算,发现了极低和非谐极低的平面内弯曲振动以及极多变的离心畸变常量。基于这些观察之前,除了计算数控键角从151到170°(取决于量子化学的计算方法),这些特征研究结果表明,乙烯ISC应该归类为有拟线性CNCSe原子链接而不是刚性的,弯曲反围平面形式。
最近,人们利用紫外光电子能谱和量子化学计算对各种不饱和或伪不饱和isc(乙烯基、2-丙烯基和环丙基isc)的物理化学性质进行了研究,以确定NCSe与不饱和基团之间的相互作用。并对相应的不饱和异氰酸酯和ITC进行了比较。通过对第一电离能的分析和比较,揭示了原子影响IE和最高占有分子轨道(HOMO)能量。即,与乙烯基异氰酸酯相比,乙烯基ITC和乙烯基ISC的IE值降低,HOMO能量增加,说明p-轨道贡献很重要,并起到减弱作用,乙烯基取代基与- NCX (X = S, Se, O)基的相互作用强度,以及降低乙烯基ITC和乙烯基ISC的第一IE。环丙基类似物也有类似的效果。特别是对于异氰酸环丙酯,由于氧原子的作用,其IE值明显高于相应的S或Se类似物。因此,这些结果表明,ITCs和ISCs之间的相互作用较弱,与异氰酸盐相比,其不饱和组分和- NCX结构。此外,在烯丙基不饱和结构中,用S或Se取代- NCX中的氧原子会导致HOMO的建立。只有在异氰酸烯丙酯的情况下,异氰酸酯基团之间的相互作用并观察到烯丙基,作者由此得出结论,必须考虑两个不相互作用的官能团。
异硒氰酸酯在癌症和其他疾病中的应用
除了有许多有用的化学应用外,一些isc也被证明具有强大的生物活性。ISC化合物已被证明对几种疾病类型有效;它们作为抗癌药物的应用已被广泛认可。这些化合物的生物活性将在以下章节中根据它们有效的疾病进行强调。
癌症。在过去的十年中,研究已经显示出各种各样的ISCs对癌症的有效预防和治疗作用。体外和体内模型均显示,与相应的ITCs相比,ISCs对癌细胞的细胞毒性更大。此外,这种巨大的治疗潜力已被证明在各种不同的癌症,如黑色素瘤,肺癌,肝癌等等。
黑素瘤。几种不同的isc已被证明对黑色素瘤模型有效。Sharma等人比较了苯基烷基ISCs的生物活性,其中苯基环和ISC片断之间的烷基链长度不同。这些化合物包括苄基异硒氰酸酯(ISC-1)、苯乙基异硒氰酸酯(ISC-2)、苯基-丁基异硒氰酸酯(ISC-4)、苯己基异硒氰酸酯(ISC-6)。在体内和体外的抗癌效果,化合物的稳定性随烷基链长度的增加而增加。具有4-和6-碳链长度的ISC-4和ISC-6被认为是最有效的化合物。因此,Sharma等人后来证明,ISC-4和ISC-6(表1)在降低UACC 903、1205 Lu、WM115黑色素瘤细胞的存活率方面比它们对应的ITCs,即异硫氰酸苯丁酯(PBITC)和异硫氰酸苯己酯(PHITC)更有效。他们也被发现在降低细胞活力方面比苯己基硒酸盐更有效(PHSC)或Akt抑制剂API-2。此外,黑素瘤细胞对ISC-4比正常的人成纤维细胞更敏感,这表明这些化合物可能对癌细胞有选择性。进一步的分析还表明,在体内外,ISC-4和ISC-6抑制Akt3信号,这一信号通路激活了约70%的黑色素瘤,并参与减少黑色素瘤细胞凋亡。这可以通过Akt和Akt底物40kda (phospho-PRAS40)中富含磷脯氨酸的Akt底物的减少得到证明。此外,无论是在体外还是在移植瘤模型中,ISC-4和ISC-6不仅抑制Akt信号通路,而且在低于相应的ITCs的剂量下诱导细胞凋亡。ISC-4后来被选作进一步研究,而不是ISC-6,因为它被认为更像药物,具有良好的LogP值(cLogP 4.4)和分子量(MW = 238),这符合利平斯基关于药物相似性的“五法则”。相比之下,ISC-6亲脂性更强,LogP值较低(cLogP 5.5)。
因此,基于这些发现,我们的小组试图阐明ISC-4对早期黑素细胞病变和正常皮肤细胞的影响。我们证明,与正常黑素细胞相比,ISC-4在诱导来自放射状生长阶段的黑素细胞病变细胞和晚期黑素瘤细胞的细胞毒性方面是2- 5倍。ISC-4可降低Akt3活性,诱导黑色素细胞病变细胞和晚期黑色素瘤细胞凋亡。此外,在没有全身毒性的情况下,在裸鼠身上局部应用ISC-4可使肿瘤体积缩小77%。同样,系统给予ISC-4可忽略器官相关毒性,提示ISC-4可能是治疗黑色素瘤的一种可行的治疗方法,且无明显的脱靶效应。
通过萘酰亚胺基取代苯环生成双拓扑异构体-萘酰亚胺异硒氰酸酯(NISC),进一步优化了苯基烷基异氰酸酯II和Akt通路抑制剂。在米托萘胺的认识基础上,一个萘酰亚胺类似物和强有力的拓扑异构酶-IIα(Topo-IIα)抑制剂, 临床试验失败了,由于系统性毒性相关的问题,我们的研究小组进行了SAR最初修改和了解ITL类似物的功能获得不同烷基链长度NITC类似物。因为ITC被证明可以减少与之相关的全身毒性问题, ITC组进一步取代ISC根是在上述研究的基础上,为了或得一个更安全,双重Topo-IIα/ Akt抑制剂。
在类似物中,具有异硒氰酸酯功能的NISC-6通过6碳烷基链与萘酰亚胺连接(表1),被确定为先导化合物。它在野生型(WT)和BRAF V600E突变型黑素瘤细胞系中同样有效,与其他类似物相比,48和72小时时细胞毒性最强。此外,尽管NISC-6在细胞毒性方面不如米托那非,但与米托那非不同的是,它对癌细胞的选择性超过了正常人类皮肤成纤维细胞(nHDF)。更一步说,硅分子对接,进一步结合体外研究,不仅表明NISC-6融入一种蛋白激酶的活性部位和拓扑酶-IIα,但也表明Topo-IIα活动抑制,以及一种蛋白激酶的磷酸化。此外,NISC-6引起的细胞死亡被发现是由于细胞凋亡的诱导。肿瘤生长在异种移植小鼠模型中,与经药物治疗的小鼠相比,黑素瘤被抑制了约69%,且未观察到明显的全身毒性迹象。因此,肿瘤抑制数据结合体外研究表明,NISC-6是一种可耐受的双作用治疗药物,具有治疗黑色素瘤的潜力。
肺癌。目前,已有两篇关于肺癌中ISCs的报道。在A/J小鼠体内的研究,基于已知的肺癌中ITCs的化学预防特性,评估了ISC-4的化学预防潜力。给药单剂量的ISC-4导致血清、肝脏和肺硒水平随时间的增加而升高,表明ISC-4在小鼠体内具有口服生物利用度。ISC-4通过抑制I期酶和诱导II期酶,调节肝脏和肺组织中I期(cyp450)和II期代谢酶的相对mRNA表达和活性水平。此外,在喂食含有ISC-4的小鼠中,使用烟草特异性前致癌物NNK处理后,观察到肝脏甲基DNA加合物的形成减少和抑制。因此,这些研究表明,ISC-4可能是一种合适的肺癌化学预防剂,因为它具有抑制致癌物代谢和增加解毒的抗启动作用。
随后的体外的SAR结果比较了不同苯基烷基ISCs对增加烷基链长度的作用(ISC-1、ISC-2、ISC-4、ISC-6;图5)在A549肺腺癌细胞中,与相应的苯烷基ITCs。与其他类型癌症的研究相比,ISCs在诱导癌细胞死亡方面比相应的ITCs更有效。Crampsie等人发现,虽然ITCs和ISCs都消耗了还原型谷胱甘肽(GSH),但ISCs比ITCs更快地完成了消耗。相比之下,ITCs的耗竭对GSH的降低作用大于ISCs。此外,与ITC类似物相比,ISCs表现出更强的氧化还原循环能力和诱导ROS水平。有趣的是,只有ITCs能诱导细胞周期阻滞,意味着细胞内的目标在S-和se -取代的类似物之间是不同的。
结肠癌。在几种不同的结肠癌细胞系中,ISC-4以剂量依赖的方式有效地抑制细胞生长。在HT-29结肠癌细胞中,发现ISC-4比其相应的硫类似物(PBITC)以及商业上可用的Akt抑制剂API-2更有效。在结肠癌的异种移植模型中,ISC-4降低了肿瘤的生长速度,但没有明显的全身毒性。肿瘤组织分析显示,ISC-4组小鼠中Akt1、前列腺凋亡反应蛋白-4 (Par-4)和磷酸化akt水平均下调。添加5-氟尿嘧啶(5-FU)治疗标准可增强ISC-4引起的生长抑制。此外,随着肿瘤抑制蛋白Par-4的加入,肿瘤生长进一步减慢。
在另一项研究中,我们评估了ISC-4的协同能力。为了观察ISC-4是否可以改善结肠癌的疗效,我们单独比较了ISC-4在RKO和SW480细胞中的活性,或者将其与19种FDA批准的抗癌疗法联合使用。在这些组合中,发现ISC-4与西妥昔单抗的组合具有协同作用。特别是,这种协同作用只在RKO细胞系中观察到,其中包含野生型KRAS:这一发现与野生型KRAS对西妥昔单抗疗效的要求是一致的。因此,在野生型KRAS细胞系中,ISC-4和西妥昔单抗协同抑制细胞增殖,其作用依赖于ISC-4的剂量,但独立于西妥昔单抗。同样,这种效应与磷酸化akt水平的降低和凋亡的增加有关。此外,在5- FU耐药结肠癌模型中,发现ISC-4和西妥昔单抗的协同作用在体内外均被保留。此外,体内研究表明,药物的组合是可耐受的,不会造成任何明显的毒性。总的来说,这些报道表明ISC-4有可能作为结肠癌治疗药物,既可以作为单一药物,也可以与化疗联合使用。
肝癌。ITC,即萝卜硫素(SFN),是Nrf2/抗氧化反应元件(ARE)信号通路最有效的诱导因子之一。在完全发展的癌症中,然而,Nrf2激活是一把双刃剑,因为Nrf2激活调节参与抗癌药物解毒的通路,可能导致癌症细胞耐药。sfn介导的这一途径导致GSH生物合成酶如限速谷氨酸半胱氨酸连接酶(GCLc)和II期解毒酶的上调。因为增强谷胱甘肽及其感应SFN的化学预防,扮演着重要的角色,由于已知化学预防 Se化合物的影响等, 假设ITC部分的硫原子与硒的替代可能使硫代嘌呤异硒氰酸酯(SFN-isoSe)(表1)具有更强的诱导ARE信号和Nrf2的能力。这一假设首先在HepG2中进行了6小时的药物治疗。而生存能力并不影响复合剂在或低于10μM,观察对细胞生存能力减少50%在SNF-isoSe的浓度为20μM时。结论是在6小时的药物暴露后,ISCs比ITCs更有效地降低细胞活力。也就是说,HepG2细胞的活力降低到对照组的0、14和0%,分别对应于sfn -异硒、苯乙基异硒氰酸酯(PEISC,或ISC-2)和苯乙基异硒氰酸酯(BISC,或ISC-1)。
类似于可行性研究,ARE诱导效应是相同的在SFN和SFN-isoSe之间,当剂量为0,5和10微摩尔时。在20μM剂量时, 由SFN-isoSe诱导荧光素酶活动水平是SFN的两倍多。与对照化合物PEISC和BISC不同,对于SFN-isoSe, ARE的增加和细胞活力的降低是剂量依赖性的。
在非癌小鼠胚胎成纤维细胞(MEF)中,无论浓度如何,SFN- isoSe诱导的核Nrf2水平也高于SFN。在MEFs中进一步的实验表明,SFN-isoSe诱导GSH的能力是依赖于Nrf2,与SFN相反,SFN显示谷胱甘肽耗竭。综上所述,这些发现表明,SFN-isoSe提高核Nrf2导致GSH酶(如GCLc)的上调,最终导致GSH水平升高。SFN- isoSe诱导GSH和SFN消耗GSH也表明,SFN- isoSe对MEFs的毒性小于SFN。因此,这些结果,与SFN- isose比SFN赋予癌细胞更大的细胞毒性相结合,表明SFN- isoSe可能是一种潜在的化学预防剂。
前列腺癌。Wu等人之前已经证明了ISC-4对其他类型癌症的疗效,他们研究了ISC-4对前列腺癌的影响。ISC-4在降低LNCaP细胞活力和诱导方面的作用是相应S类似物PBITC的4倍。与早期报道的结肠癌和黑色素瘤的研究结果相反,ISC-4并没有抑制Akt的磷酸化,这说明在LNCaP前列腺癌细胞的细胞毒性机制中,Akt的抑制并不是导致ISC-4磷酸化的原因。相反,ISC-4在转录后可降低雄激素受体(AR)和前列腺特异性抗原(PSA)水平,与细胞凋亡无关。
其他实验表明,ISC-4诱导ROS,通过激活p53信号,不仅抑制AR/PSA,而且促进细胞凋亡。特别是p53上调凋亡调节剂(PUMA)-Bax (bcl -2相关的X蛋白)的固有凋亡级联反应被激活。p53的缺失导致了凋亡的衰减,这一效应与ISC-4对AR/PSA的影响以及p53下游靶点的下调无关,也能减弱isc -4介导的细胞凋亡。
乳腺癌。Cierpia?等人合成一系列有机氟ISC萝卜硫素类似物与不同的化学成分,并比较他们的抗癌活动相对于ITC萝卜硫素。由于SFN已知可诱导乳腺癌细胞周期阻滞和细胞死亡,因此在不同的乳腺癌细胞系中筛选了类似物。通过几项测试,发现新的类似物相对于SFN,对癌细胞生长有更强的选择性抑制作用。
在各种类似物中,4-异氰基-1-丁基4 ' -氟代酶亚砜(化合物8i)(表1)被认为是潜在的先导化合物。尽管它在两种被测试的乳腺癌细胞系(MDA-MB-231和MCF7)中都有效力,但它对与激素无关、三阴性的MDA-MB-231细胞选择性最强。MDA-MB-231细胞是一种侵袭性乳腺癌的模型,对传统的、靶向的或基于激素的疗法无效。此外,化合物8i对癌细胞总体上具有选择性:在正常细胞系中,化合物8i的毒性比SFN小,在癌细胞中,化合物8i的细胞毒性比SFN更强。
传染病疟疾。Nieves等人合成了几种基于(−)-8,15-二异氰酸酯-11(20)-亲和素的结构的化合物,亲和素是一种已知的具有有效的抗疟疾和抗细菌作用的代谢物,从海洋海绵Svenzea flava中分离得到。之前SAR研究表明,异氰酸酯的功能是生物活性所必需的,其位置对活性也很重要。记住这一点,根据观察海绵状异氰酸酯-、ITC-和甲酰胺-二萜类化合物也具有生物活性,其新型的二萜类化合物在不同的位置加入了ISCs和ITCs作为潜在的药效基团来测试。在两个不同的恶性疟原虫疟原虫系(氯喹-)中类似物的筛选表明敏感的3D7和耐药的Dd2)显示所有的ISC类似物都表现出亚微摩尔的抗疟原虫活性。这些混合的化合物,化合物5(表1),其中包含两个ISC,比标准更强有力的药物氯喹Dd2 (IC 50 0.0066μM相比0.0519μM)。此外,它对耐药株表现出更大的选择性,与氯喹(SI =4518)相比,具有更高的选择性指数值(SI = 7356)。
肺结核。在上一节所述的同一研究中,Nieves等人也筛选了他们的ITC-和ISC-amphilectane二萜来对抗Mtb h37 Rv,以阐明最低抑制浓度(MIC)。经isc功能化的杂化化合物MIC值最低;特别是,混合化合物5和6的最低抑制浓度为 3.9和2.1μM。相比之下,ITC-功能化的 amphilectane二萜有更低的MIC,从26.8到99.1μM。此外,在杂种化合物5和6中,化合物6被认为是最有效、最具选择性的,这一发现,与ITC -杂交类似物的数据相结合,再次表明硒在这些化合物的选择性和强大的细胞毒性中发挥作用。
利什曼病。由于硒化合物和抗肿瘤药物均表现出利什曼杀菌活性,因此,我们研究了ISC衍生物在大利什曼原虫和亚马逊利什曼原虫原体和胞内无利什曼原虫形态中的抗增殖活性。在检测的类似物中,发现NISC-6具有抑制利什曼原虫原核细胞增殖的作用,其纳米范围的IC 50值是参考药物两性霉素B的3倍(两性B)。此外,NISC-6的选择性指数最高(SI = 416.7),几乎是参考药物的10倍。虽然L. amazonensis对Ampho B和NISC-6的敏感性较低,但NISC-6的SI (SI = 41.7)仍然是Ampho B的2倍(SI = 20.5)。NISC-6也被证明能引起细胞周期阻滞,证据是复制相关基因如增殖细胞核抗原(PCNA)、拓扑异构酶-2 (TOP2)和小染色体维持复合体4 (MCM4)的mRNA水平下降。此外,与未处理的细胞相比,NISC-6处理导致G1期寄生细胞数量增加,S期减少(表1)。
结论和观点
从前面几节可以明显看出,ISCs在一些化学学科中显示出了很大的应用潜力,如理论、计算、有机金属、聚合物和无机化学(图4)。
然而,在这些用途中,由于缺乏有效的、长期的治疗以及预期的癌症病例和死亡的增加,作为抗癌剂的应用尤其令人感兴趣。此外,所述的研究表明,ISCs是有效和有效的抗癌和化学预防剂,在大多数情况下比相应的ITCs更有效。
通过增加与苯基烷基异氰酸酯(ISC-1、ISC-2、ISC-4、ISC-6)的烷基链长度,桥接苯基环和ISC片断,增加了体外和体内的生长抑制力(图5)。这一发现与以往各种苯基烷基ITCs的体外研究结果相似,即随着烷基链长度增加至8个碳,其效价增加,但与体内研究不同,在体内研究中,烷基链长度增加并不对应体内更强的肿瘤抑制作用。重要的是,在isc研究中,观察到对照硒代半胱氨酸缺乏疗效,这表明单硒并不是iscs增强生长抑制作用的原因,而是isc结构与硒的结合。还推断,较长的烷基链长度可能有助于ISC或ITC基团与酶活性所需的关键残基的接触和结合。
然而,在NISC类似物的情况下,增加的烷基链长度并不总是对应更强的生物活性,尽管确定NISC-6是与6碳烷基链(测试中最长的烷基链)最有效的化合物。这与相应的硫类似物形成对比,硫类似物中增加的烷基链长度增加了效价。此外,不像硫类似物NNITC-2,其中硝基取代萘酰亚胺环系与未被取代的nnc -2相比,NNISC-2的细胞毒性较低,表明在Se类似物的情况下,硝基取代是不利的(图5)。
对于SFN的有机氟ISC类似物,硫原子的氧化,当与4-氟苯环和亚砜基团之间的亚甲基基团结合时,发现增加了抗癌效能。此外,增加硫基官能团和ISC片断之间的烷基链长度(从4个碳到5个碳)会导致对正常细胞系的细胞毒性增加,从而降低化合物的癌细胞选择性(图5)。
根据对这些SARs的观察,可以得出结论:氧化态、环取代和烷基链长度对ISCs的效能和效能有影响。同样,这些因素都有助于这些化合物的药物相似性,这是一个必须考虑的性质,以优化和提高这些化合物作为潜在的抗癌疗法和化学预防剂。因此,在上述的一些SAR研究中已经考虑了药物相似性。例如,计算分析表明,NISC-6符合Lipinski’s Rule 5的原则,其计算的拓扑极性表面积预测具有较高的口服生物利用度。此外,使用苯基烷基ISCs, Sharma等人注意到在苯环和ISC之间增加烷基链的长度的功能增加了ITCs和ISCs的亲脂性和CLogP值,但对ISCs的亲脂性和CLogP值的影响更大。随着烷基链长度的增加,化合物应能更容易、更有效地扩散到细胞内。虽然ISC-4和ISC-6同样有效,但只有ISC-4的CLogP符合Lipinski的五项规则。因此,这一知识,以及已知的诱导食道癌变的大鼠由于PHITC的高亲脂性使ISC-6成为不太适合的抗癌和/或化学预防剂。
因此,这些观点又回到了另一个重要的问题:为什么要使用硒?硫和硒是元素周期表上同一家族的两个成员,它们的氧化态相同,这两种元素之间有什么不同之处,使得它们具有更强的抗癌功效和效力?SARs表明,在体内外,ISCs比ITCs具有更强的活性,并具有更强的亲脂性。也许这些生物活动的差异可以根据它们的原子和电子特性而不是仅仅根据它们的分子特性来解释。虽然已知S和Se具有相似的电负性值,但它们的原子、离子和范德华半径以及它们的氢键能力不同。与S相比,Se具有更大的原子、离子和范德瓦尔斯半径,Se原子上的电荷更分散,这削弱了分子间的相互作用。硒有一个比硫更强的π-bonding,此外,虽然S有时被认为是氢键的受体,与Se的氢键可以忽略不计。也许,当整个分子与相应的靶蛋白相互作用时,这两种元素之间的细微差别组合在一起,就可以解释所观察到的抗癌功效和效力的差异。
如前所述,Crampsie等人试图从生物学机制上阐明苯烷基ITCs与ISCs中S和Se的区别。拟一阶动力学实验表明,无论是ITCs还是ISCs,与GSH的反应速率均随烷基链长度的增加而降低。虽然ISCs的反应速度比ITCs快,但在S和Se类似物之间,GSH的平衡浓度没有显著差异。在肺癌细胞中,GSH在ISCs中消耗得更快,但在ITCs中,总消耗更大。也许ISCs更强的反应性可以归因于Se的价电子比硫更不稳定,因为它的C=Se键不如C=S稳定,使得它对巯基亲核试剂的反应更强烈。
与报道的ITCs相似,ISC化合物与亲核试剂的反应活性表明它们在体内系统的整体稳定性较差,从而影响了它们的类药物特性。由于ITC功能的亲电性,天然ITC SFN的半衰期仅为1.77±0.13 h。然而,由于硫醇共轭代谢物ISC和ITC的化合物,与蛋白质反应形成硫醇(如谷胱甘肽、半胱氨酸和防治作用)或在硫醇尿酸形成通路,也被证明是有效的抗癌药物,这些化合物可以一起持续比较久活性代谢物,从而导致异硫氰酸酯和ISC的功效。然而,可以通过创造更稳定的水溶性前药来克服稳定性差的限制,如Jiang等人最近报道的,SFN将在未来创造临床相关的ISC候选药物。
此外,ISCs的氧化还原循环生成ROS的效率更高,范围也更广,这与−SeH的氧化还原循环和反应活性高于−SH的事实一致。因此,这些数据表明,ISCs和ITCs的细胞毒性作用与巯基相互作用有关。尤其是isc,增强细胞毒性效应被认为是由于谷胱甘肽耗竭和氧化还原循环,而与谷胱甘肽反应的速率无关,由于数据反驳了原先的假设,即用Se取代S会降低碳原子的亲电性,从而降低硫醇的反应活性。
无论如何,isc已显示出作为潜在癌症治疗或化学预防剂的巨大潜力。除了单纯的ISC-4和西妥昔单抗外,未来癌症相关的ISC研究应该寻找其他潜在的联合治疗方法。例如,可以双重Akt Topo-IIα抑制剂与BRAF V600E抑制剂PLX4032 NISC-6协同作用,帮助延缓其在黑色素瘤中的耐药性?当有机氟ISC萝卜硫素类似物与标准化疗联合使用时,三阴性乳腺癌的侵袭性是否可以降低?
此外,所述iscs可以类似于n isc-6的方式进行修饰,其中isc部分和必需基团与已知抑制剂和化疗剂的生物活性功能结合,并且可以将所述修饰后的化合物的生物活性与其各自的母体化合物进行比较。此外,这些iscs的反应性可在并入几个isc和/或itc部分时进行研究。此外,胶质母细胞瘤是一种侵袭性癌症,没有有效的isc类似物。未来的研究还可以集中在合成能够跨越血脑屏障的有效、靶向ISC类似物上。
正如生物学研究所提示的,要获得一种非常有效的、具有癌症细胞选择性和生物可利用性的ISC,需要在优化亲脂性(即、氧化态和/或芳香环取代物而不丧失已知的生物活性。为了获得这些优化的ISCs,还必须有可靠、简单、安全的合成方法。虽然所述的一些合成方法有其局限性,但未来的研究可以努力实现更绿色的方法,以产生所需的isc,并获得良好的产量。这种合成方法的发展将加速这些化合物的结构优化。
除了抗癌的ISCs外,强调ISCs在其他疾病中的作用也很重要。Nieves等人指出,新的ISCs正在作为抗疟疾和抗结核药物开发。通过这些发现的扩展,未来的研究可以集中于开发用于其他传染病的ISCs,如霍乱或脑膜炎。在现有抗菌药物中添加ISC基团,抗菌药物的生物活性部分可与现有的ISC化合物融合,与多种抗感染药物联合使用可评估ISC的协同作用。
作为一个总结,ISCs有能力在未来几年形成许多化学领域,如理论、有机金属、聚合物和无机化学,这已经被他们的研究证明了。它们在药物化学领域以及在癌症和人类疾病方面尤其有前途,因为它们已被证明比同构硫类似物更有效。随着更多的研究和结构优化,这是一个很大的希望,新的ISC化合物以及一些在这篇综述中描述的ISC化合物可以推进到临床试验,并最终在临床作为新的癌症药物和化学预防剂。
本文由福山生物整理翻译,转载请注明出处。