






最新资讯



-
电话: 400-113-6988 邮箱: dongfangxicao@163.com 地址: 深圳市南山区粤海街道高新中三道9号环球数码大厦19楼
↓ 微信扫码识别关注我们 ↓ 
四唑类有机硒化合物的硅、分子和生化组合合成及其对肝癌的选择性研究
发表于:2020-06-01 作者:admin 来源:本站 点击量:16718
四唑类有机硒化合物的硅、分子和生化组合合成及其对肝癌的选择性研究
(Combinatorial synthesis, in silico, molecular and biochemical studies of tetrazole-derived organic selenides with increased selectivity against hepatocellular carcinoma)
DOI: 10.1016/j.ejmech.2016.06.005
(Combinatorial synthesis, in silico, molecular and biochemical studies of tetrazole-derived organic selenides with increased selectivity against hepatocellular carcinoma)
DOI: 10.1016/j.ejmech.2016.06.005
摘要
采用叠氮-尿苷(azido-Ugi)和顺序亲核取代(S N)策略合成了新型四唑基二硒醚和硒醌。通过对哺乳动物TrxR1的分子对接研究,预测了新合成化合物的抗癌潜力。使用肝细胞癌(HepG2)和乳腺癌(MCF-7)癌细胞对化合物的细胞毒性活性进行评价,并与它们在正常成纤维细胞(Wi-38)细胞中的细胞毒性进行比较。使用2,2-二苯基-1-吡啶酰肼(DPPH),谷胱甘肽过氧化物酶(GPx)样活性和博莱霉素依赖性DNA损伤来评估合成化合物的相应氧化还原性能。一般而言,二硒类化合物与MCF-7细胞相比,对HepG2具有优先的细胞毒性。与依布硒啉相比,这些化合物还表现出良好的GPx催化活性(高达5倍)。选择硒醌18、21、22和23分别检测caspase-8、Bcl-2和Ki-67的表达水平,有趣的是,这些化合物下调了Bcl-2和Ki-67的表达水平,并且与未经处理的HepG2细胞相比,激活了caspase-8的表达这些结果表明,一些新合成的化合物具有抗HepG2活性。
1.简介
面向多样性的合成(DOS)已经成为制药工业中一个强大的工具,现在经常用于药物发现,但主要用于使用集中的文库来改善所需的性能。在这种情况下,可以通过筛选不同骨架的小分子来确定和优化特定的生物靶点。近年来,新的DOS途径不断发展;然而,多组分反应(MCRs)是广泛研究的合成策略之一,它可以有效地生成小的化学实体,增加分子的多样性和复杂性。幸运的是,基于异氰酸酯的多组分反应(IMCRs)是MCR最常见的子类,它提供了一种简单的方法,可以在单一步骤中引入结构多样性和分子复杂性。在所有已知的IMCRs中,帕西尼反应和Ugi反应已经成为一种强有力的手段,通过提供深度肽、n -甲基肽和肽类(包括二聚体)来构建肽样化合物。
尽管传统的、基本形式的Passerini和Ugi产物具有潜力,但它们的性质限制了它们的发展;即,酯基和/酰胺基结构。因此这些化合物;容易代谢,即在体内它们可能被裂解或修饰成潜在的或多或少的活性或理想的代谢物。相应地,将Passerini和Ugi反应的潜势扩展到获得更多受限的支架,即杂环结构。这就是Groebke和azido-Ugi反应发挥作用的地方。这些反应可或得生物活性咪唑和四唑类化合物,限制基序。
另一种获得更多多样性和复杂性的方法是用后续的缩合修饰来标记Ugi反应,通常是在初始Ugi产物的主干上附加正交官能团。双官能团的结合允许随后的二次转变(如亲核取代(S N) 反应、环缩合和环加成),并达到第二层次的多样性。鉴于前者,已经报道了Ugi反应与其他反应的顺序耦合不同的修饰后反应如Heck、Diels-Alder、PicteteSpengler、Petasis、Mannich、Wittig和Click等反应合成了多种重要的杂环类支架,支架具有密集的结构特点和功能。这种双层的方法非常重要,现在它经常用于药物发现。
最近,我们采用多组分策略(如Passerini, Ugi和Ugi / S N,迈克尔反应)合成混合结构含有不同硒有机库耦合tobioactive药效团(例如,醌类、萘、环胺)或药物相关杂环化合物(如thiazolidinone,吡唑和thiazolopyrimidine)。其中一些化合物在亚微摩尔浓度下对各种类型的癌细胞表现出细胞毒性,如肝细胞癌(HepG2)、乳腺腺癌(MCF-7)、A-498(人肾癌)和A-431(人表皮样癌)细胞系。值得注意的是,HepG2和MCF-7细胞的毒性更明显。此外,其中一些化合物在与正常细胞如HUVEC(人脐静脉内皮细胞)、WI-38(人肺成纤维细胞)和HF(人原代成纤维细胞)细胞系进行测试时显示出较低的细胞毒性。潜在的细胞毒性和选择性机制非常有趣,因为这些化合物可以作为抗氧化剂或促氧化剂,依赖于它们的氧化还原特性和它们所处的细胞内氧化还原环境。
在正常细胞中,有机硒化合物作为抗氧化剂,从而保护细胞免受氧化损伤。另一方面,这些化合物成为氧化应激细胞(如MCF-7和HepG2)的促氧化剂。这一双峰功能提出了有机硒化合物不仅作为化学预防剂,而且作为选择性化疗药物。
虽然我们只能推测其可能的作用模式,但它们的细胞毒性主要归因于caspase 3/7的激活和随后的诱导细胞凋亡。此外,还观察到不同的表型变化,包括内质网、肌动蛋白细胞骨架和细胞形态改变以及细胞周期阻滞和各种生化变化(如活性氧和谷胱甘肽水平)。有趣的是,我们发现以硒为基础的醌类是最活跃的化合物之一,具有增强的抗癌活性。
为了继续我们的项目,开发有治疗前景的有机硅制剂,我们在此报告了通过叠氮- ugi和叠氮- ugi /S N方法学,开发出一种简便的方法来获得对称的二硒醚和硒醌类四唑类化合物。新合成的化合物各自的作用模式通过两方面进行评估:使用HepG2、MCF-7和正常细胞(WI-38)进行细胞试验,研究它们相应的细胞毒性,并估计它们对caspase-8、Bcl-2和Ki-67分子生物标志物表达水平的相应影响;b)利用DPPH、GPx-like活性和博莱霉素依赖DNA损伤实验,探索合成化合物的氧化还原调节活性。此外,在硅分子模型研究中,包括场对齐和对接研究,将作为一个初步的预测工具来估计化合物的抗氧化和细胞毒性特性。
2.结果与讨论
2.1设计与合成
直到最近,合成有机硒化合物还不是一项容易的任务,包括使用昂贵/有毒的起始材料。近年来,在硒杂环化合物、硒氰酸盐、硒化物和二硒化物等不同类型有机硒化合物的合成方面取得了显著进展。
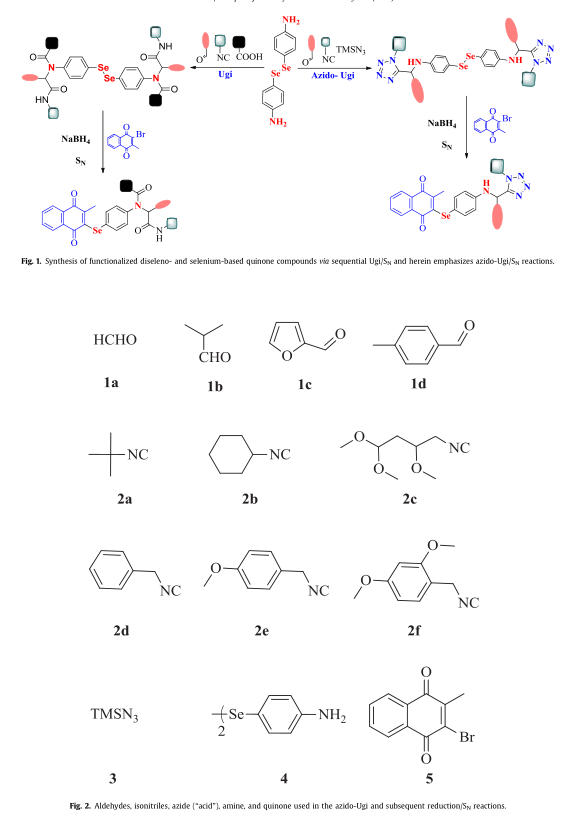
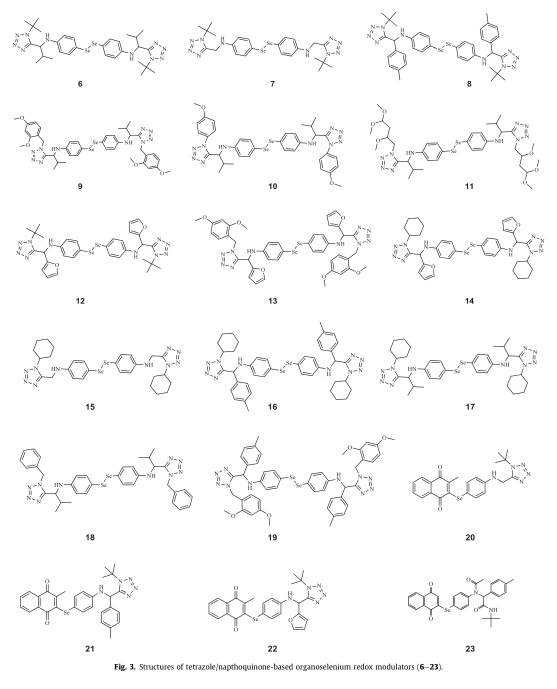
作为我们旨在开发有机硅类化疗药物的项目的一部分,我们报告了通过叠氮唑- ugi和序贯SN策略合成了四唑类对称二硒醚和硒醌类化合物(6-22)(图1)。
基于我们小组最近的发现,我们设想四唑基的有机硅支架可能是更有效的抗癌药物。四唑作为几种生物活性化合物和许多已上市药物(如戊四唑、西洛他唑、头孢替唑、厄贝沙坦、氯沙坦)的核心骨架,在制药市场受到了相当大的关注。此外,这些化合物具有特殊的意义,因为四唑是羧酸基(COOH)的生物同工酶,但具有更好的药效、良好的理化性质、更好的药代动力学和代谢稳定性。这主要是由于四唑更大的尺寸和更优越的亲脂性(z10倍多),进而导致底物受体相互作用的增加。
构建四唑环体系的方法很多;然而,azido-Ugi反应在自动化、反应时间和总收率方面都优于经典方法。为了保证第二级多样性,4,4 0 -二苯胺(4)被用作双功能键合子,因为它拥有一个便于叠氮唑-ugi反应的氨基以及一种易于释放的亚硒酸盐亲核试剂(通过NaBH4还原二烯酰胺在原位形成),适用于后续的SN反应。
在文库构建方面,有6种结构多样的异氰基(如叔丁基异氰酸酯(2a)、环己基异氰酸酯(2b)、4-异氰基甲基丁烷-1,1,3-三醇(IPB, 2c)、苄基异氰酸酯等。4-甲氧基苯并异氰酸酯(2e)和2,4-二甲氧基苯并异氰酸酯(2f)被用于文库验证,部分原因是它们有可能在ugi后发生其他反应。以脂肪族(多聚甲醛(1a)、异丁醛(1b)、芳族(4-甲基苯甲醛(1d)、糠醛(1c))为氧组分,以三甲基硅氧烷(TMSN3) 3为酸组分(见图2)。
四唑基对称二硒醚类化合物(6-19)的合成方法是在4(1个当量)的甲醇溶液中加入2个当量的醛1,然后加入2.5当量的TMSN3和异氰酸酯2。反应完成后,二硒酰胺(6-19)与NaBH4还原得到相应的亚硒酸钠,与2-溴-3-甲基-1,4-萘醌(5)通过SN反应(或添加/消除序列)得到四唑基硒醌类化合物(20-22)。
值得注意的是,该反应序列也可以在一个锅(五种成分)中进行,而无需分离最初的叠氮唑- ugi产物。总的来说,6-22的合成进展顺利,化合物得率极好(高达96%,图3)。
2.2 生物活性的硅预测
分子模拟技术在抗肿瘤药物开发中具有重要的应用价值。因此,他们被用来虚拟评估氧化还原调制和新合成的化合物在生物筛选前的潜在抗癌特性。采用现场比对的方法进行了三维相似度研究二硒类化合物和两个已知的具有类似gpx活性的相关参考化合物。此外,还在硫氧还蛋白还原酶(TrxR1)[48-50]的二聚界面域进行了对接研究,以探索这些化合物作为TrxR1抑制剂的能力。
2.2.1 3D相似度研究
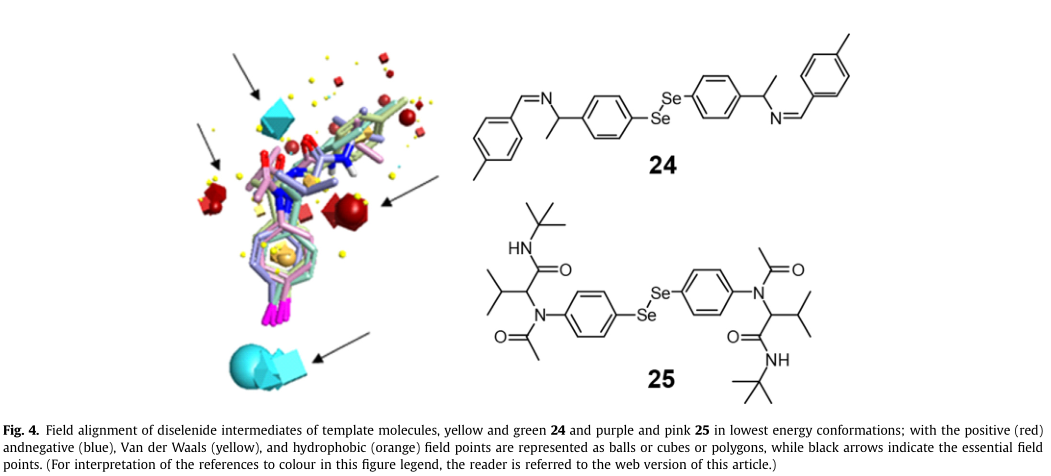
对对称二硒醚类化合物进行了三维相似性研究,以确定这些化合物是否具有与已知GPx模拟活性的参考化合物相似的静电模式、构象和/或形状。化合物(Z)-N-(4-甲基苄基)-1-(2-((2-((1-((E)-4-甲基苄基氨基)乙基)苯基)苯基)乙胺(24)和2,2’ -((二硒(4,1-苯基))双(乙酰氮杂二烯))双(叔丁基)-3-甲基丁酰胺)(25)与公认的GPx模拟活性,用作模板和比较的目的。场对齐技术用于根据分子场对分子进行对齐。与24和25场模式相似的分子有望具有相似的生物活性模式,特别是在氧化还原反应中。随着分子相似性趋近于一,排列和静电结构的相似性越好。
由于缺乏24和25的生物活性构象,使用Forge 10.4的field Templater模块主要生成一个field模板随后,在Forge 10.4中,我们的二硒化物的硒醇形式通过场对准模块与模板进行了场对准。
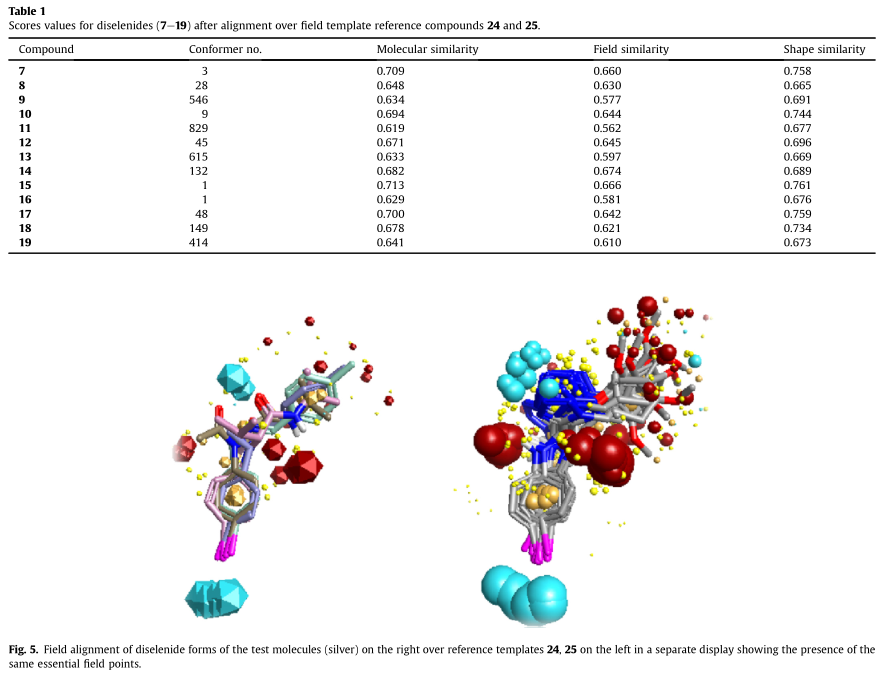
化合物24和25的硒醇形式被加载为单一的2D结构,然后传递信息生成一组不同的构象。在探索的一组分子构象空间中的公共场模式选择排名第一的字段模板(分数:分子相似性:0.713,字段相似性:0.656,原始字段得分:-67.014,形状相似性:0.769,原始形状得分:154.4)生成的场模板显示了硒醇基团周围的负场点(蓝色),而对位取代区周围有多个正场点(红色)化合物25的构象显示了一些额外的负场点(图4)。
应用场对齐后,在成对匹配的基础上选择每个测试分子的最可能构象。平均分子相似性约为0.619-0.713。对生成的模板比对后,化合物的得分值如表1所示。我们的测试化合物中也有必要的场点(图5)。因此,场对齐技术表明,我们的新二硒醚类化合物与参考化合物24和25具有相同的静电模式,至少在更稳定的构象e形状上是相同的,这表明它们具有类似的GPx模拟活性。
2.2.2 对接研究
许多有机硒化合物被报道可抑制某些癌细胞的硫氧还蛋白还原酶(TrxR),从而激活凋亡通路。为了预测我们化合物的抗癌活性和凋亡能力,我们与哺乳动物TrxR酶进行了对接。TrxR1为四种单体的均二聚四元结构。每个单体主要有两个区域:n端FAD和NAD结合域和柔性c端二聚界面域。
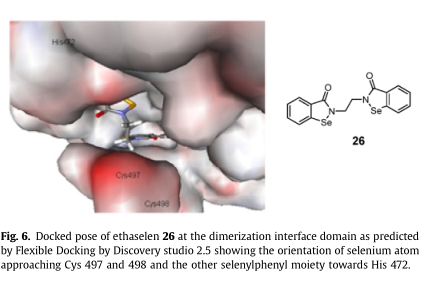
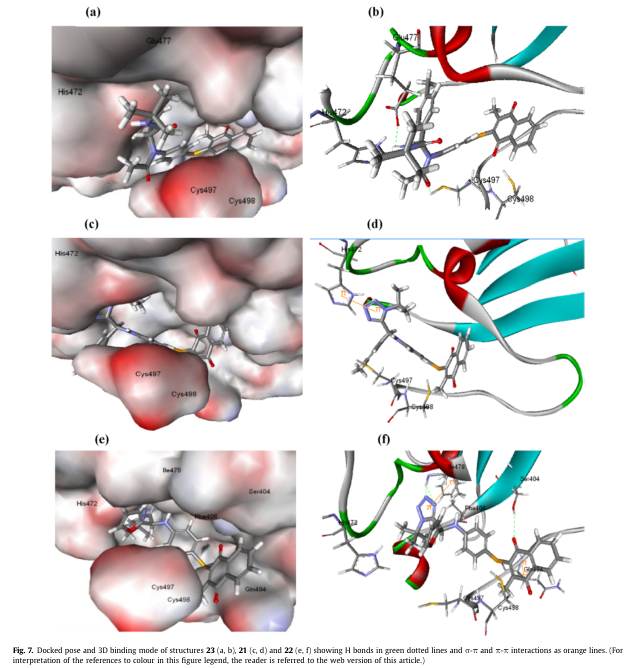
Wang等人报道了有机硒化合物依布硒啉 26与哺乳动物TrxR1相互作用机理的硅内模拟模型他们提供了一个基于证据的解释,即依布硒啉26靶向哺乳动物TrxR1在二聚界面域的C末端活性位点,分别与蛋白质的Cys497和Sec498氨基酸形成两个分子间的硒烯基硫化物(Se-S)和硒烯基硒化物(Se-Se)共价键;因此它起着一种不可逆的酶抑制剂。
由于缺乏人TrxR1三维结构复合物与抑制剂聚合,采用柔性对接协议进行柔性对接。这个协议允许一些受体柔性配体对接时的灵活性。指定氨基酸的侧链可以在停靠期间移动。这使得受体能够适应不同的配体在一个诱导契合模型中。
依布硒啉26成功对接,选择了理想的位置,即将硒原子指向相互作用的半胱氨酸残基497和498,同时将另一个硒苯基的苯基指向其472(图6)。这种位置与分子动力学模拟研究中报道和提出的位置相似。
此外,与0.75 kcal/mol的依布硒啉26相比,化合物21和22的结合能也更高,分别为7.07和13.92。另一方面,化合物18表现出最低的结合能(见补充材料)。
结构21和结构22的结合模式显示了His 472或Phe 406的芳香环与四唑环之间的p-p相互作用(图7d和f)。此外,22对接位置还显示了额外的相互作用:gln494和醌环之间的δ-π相互作用,Ile 478和四唑环上各2.3 A的氮(N3和N4)之间的两个氢键,以及Ser 404和醌的羰基氧(2.5 A)之间的一个氢键。这些相互作用将化合物21和22锚定在结合位点上,使得硒原子以6.8 a和5.8 a的距离更接近498半胱氨酸残基。
2.3 药理学,毒性和抗氧化剂概况
2.3.1氧化还原活性化合物对人细胞系的细胞毒活性
为了检查这里的化学发展是否已导致潜在有趣的候选癌症治疗,我们的初步目标是检查是否新合成的化合物具有任何抗癌活性的预测,在硅的研究。因此,我们对两种哺乳动物癌细胞株HepG2和MCF-7细胞进行了细胞毒性筛选。基于之前的广谱筛选,选择了这些细胞,结果表明,在MCF-7和MCF-7的情况下,有机硒化合物的毒性通常更明显。此外,这些细胞系是相当合适的药物靶向模型。另一方面,WI-38正常肺成纤维细胞也被用来初步了解这些化合物的选择性细胞毒性,并缩小最有趣的化合物的数量,以便进一步深入研究。
将细胞与不同浓度的测试化合物孵育48小时,使用标准的颜色-imetric3-(4,5-二甲基噻唑-2-yl)-2,5-二苯基四唑溴化铵(MTT)法定量测定代谢活性细胞的数量,从而估计其生存能力。5-FU作为阳性对照,因为它通常用于辅助和姑息性癌症化疗。根据各自的剂量反应曲线估计细胞毒性,并以化合物浓度表示,该浓度使处理细胞的吸光度降低50% (IC 50),即50%的生长抑制。治疗指数(TI)被定义为抑制50%正常WI-38细胞生存能力的化合物浓度与抑制50% HepG2细胞生存能力的化合物浓度之比(表2)。
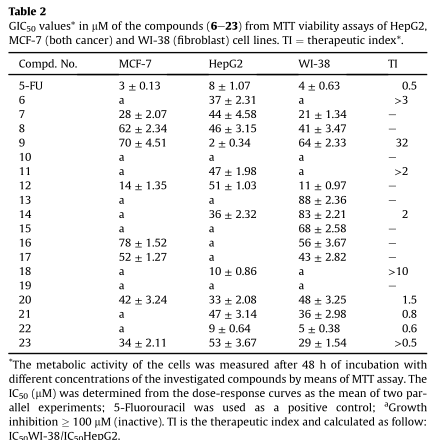
有趣的是,在正常(WI-38)和癌症HepG2细胞之间观察到明显的毒性区差异。因此,TI的计算结果与HepG2细胞一致(IC 50WI-38 /IC 50HepG2)。化合物9和18被发现是最敏感的(各自的选择性指数¼32和11个)。此外,化合物6、11、14和20的Ti值高达3,反过来也高于标准基准化合物5-FU。
当然,这些最初的研究不能直接转移,因此需要更多的研究,使用更多的正常细胞和肿瘤细胞,并最终进行有机实验。
2.3.2 HepG2细胞中caspase-8、Bcl-2和Ki-67分子标志物的评价
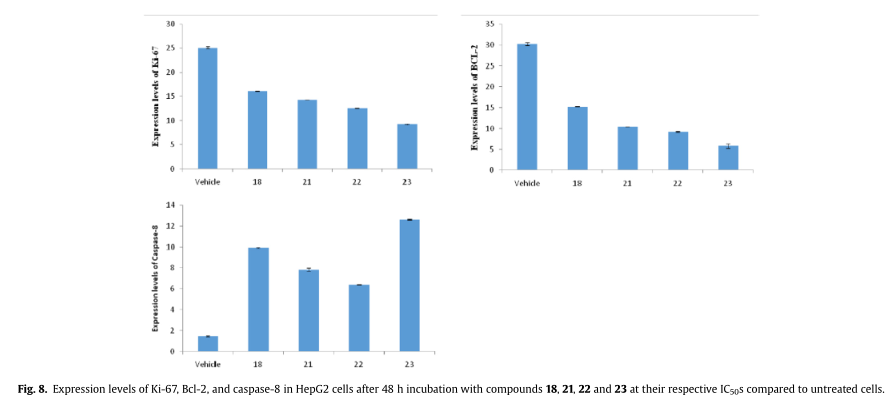
为了进一步研究可能涉及的信号通路,并在分子水平上获得关于最有希望的化合物18、21、22和23的作用模式的线索,检测HepG2细胞中肿瘤增殖、抗凋亡和凋亡蛋白标志物的表达水平。鉴于前者,我们选择了Ki-67增殖标记物而促凋亡的caspase-8和抗凋亡的Bcl-2蛋白则被用来监测凋亡诱导。
如图8所示,与未处理的细胞相比,化合物18、21、22和23能够上调caspase-8的表达,下调Ki-67和Bcl-2的表达。值得注意的是,在测试化合物的化学结构和相应的表达调节活性之间存在明显的相关性,表明它不是一般的硒的细胞毒性作用。毫不奇怪,与TrxR1对接研究一致,硒醌21、22和23比对称二硒醚类更活跃。有趣的是,23在奎奴亚胺类似物中表现出优越的活性,这值得进一步研究。这些结果与我们之前的研究一致,其中硒基醌类诱导了一系列的生化改变,其中包括细胞caspases的激活和凋亡的诱导。这些变化似乎主要发生在特定的细胞(如HepG2)中,细胞内氧化还原平衡受到干扰。
虽然现在解释化合物23为什么是这些测定中最活跃的物质还为时过早,但人们可以推测,这种化合物可能击中不止一个特定的细胞靶点,并引起广泛的蛋白质和酶的修饰,使之活化。此外,23也可能被细胞吸收并在体内被修饰成活性代谢中间体。由于这些都是未知的,推测其确切的代谢、动物体内的药代动力学、特定组织的富集或降解的细节还为时过早,尽管这些问题显然很重要,将成为我们未来研究的一部分。最终,由于这些化合物的结构提供了相当大的修饰余地,而现在合成衍生物也很简单,这将成为一个结构变异的未来研究的起点。
2.3.3 抗氧化活性评估
近年来,有机硒化合物因其具有抗肿瘤的化疗潜力而受到广泛关注,特别是在公共卫生和制药行业。因此,对这类化合物提出了不同的作用模式。然而,活性氧调节已被认为是有机硒化合物的一个重要标志性机制。
由于有机硒化合物有时被认为是氧化还原的调节剂,因此对合成的化合物的氧化还原活性作了进一步的估计。采用不同的生化测定方法,如DPPH、gpx类活性和依赖博莱霉素的DNA损伤测定。氧化还原状态在对抗和区分肿瘤细胞与正常细胞中至关重要。
2.3.3.1 DPPH自由基清除实验
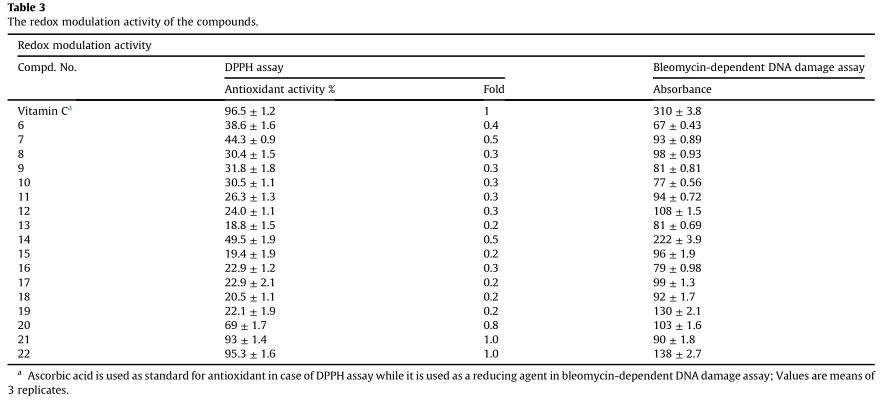
评价有机化合物抗氧化性能的方法很多;然而,体外DPPH化学分析由于其简单、快速而在体内相关性有限,常被用于评价有机物和营养制品的自由基清除活性。化合物的抗氧化活性是通过其降低DPPH的能力来估计的。自由基(甲醇中的紫色)生成DPPHH(颜色-)和相应的自由基清除活性是通过517 nm处吸光度的降低来评估的。以标准水溶性抗氧化剂维生素C为阳性对照。
如表3所示,四唑-硒醌类化合物20和21是本实验中活性最强的化合物,与抗坏血酸相比,它们具有清除自由基的活性。这与我们之前的报道一致,醌类是低浓度的抗氧化剂。
2.3.3.2博莱霉素DNA损伤试验
采用博莱霉素-铁DNA损伤实验,评价合成化合物的促氧化活性。建立了一种准确而独特的检测药物和食品抗氧化剂潜力的方法。博莱霉素抗菌素具有很好的抗癌作用通过二价铁离子辅助的DNA损伤。博来霉素形成一个复杂的二价铁离子,进而催化DNA降解。总之,助氧化剂化合物减少bleomycin-Feþ3,bleomycin-Feþ2,在有氧条件下,从而诱导DNA降解,可随后spectrophotometrically吸光度的增加在532海里(表3)。在这个试验,维生素C作为还原剂和减少bleomycin-Feþ3复杂诱导DNA降解。简言之,亲氧化剂化合物在有氧条件下将博莱霉素铁蛋白3降低到博来霉素铁蛋白2,从而诱导DNA降解,随后在532 nm处增加吸光度(表3)。本实验以维生素C为还原剂,还原博来霉素三价铁离子络合物,诱导DNA降解。
用改进的方法,随着吸光度的增加,更多的博来霉素三价铁被转化成博来霉素二价铁,从而诱导DNA降解,从而支持这些化合物的氧化活性。化合物6、9、10、13和16抑制了博莱霉素三价铁的还原,从而减少了博莱霉素二价铁的生色形成,保护了DNA另一方面,化合物14、19和22显著地诱导了DNA降解,比其他被研究的化合物更多(表3)。
根据活性氧调节理论,解释了有机硒化合物的细胞毒活性。化合物22主要通过其明显的促氧化特性发挥其细胞毒性作用。另一方面,其他化合物参与细胞毒活性可能存在其他机制,有待进一步研究。
2.3.3.3。谷胱甘肽过氧化物酶活性测定。
硒是多种酶的重要组成部分,包括GPx和硫氧还蛋白还原酶。这些酶大多含有作为亚硒半胱氨酸氨基酸的硒辅因子,而亚硒半胱氨酸是其相应的抗氧化特性的来源。自从发现有机硒化合物作为良好的GPx模拟物,这些制剂中有许多已经被开发用于控制与氧化应激相关的各种疾病,如癌症、阿尔茨海默病和炎症性疾病。
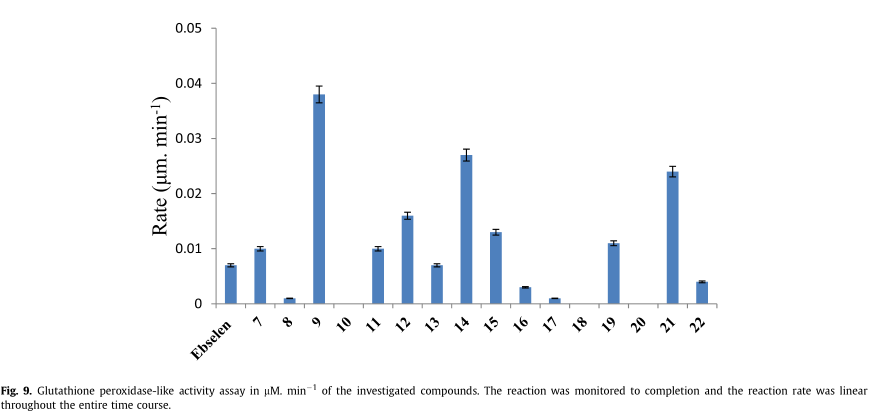
用NADPH还原酶联用法测定了新化合物的GPx样活性。该方法常用于评估有机硒化合物的类GPx活性。通过GPX还原过氧化物,随后将氧化谷胱甘肽(GSSG)转化为还原形式,并将NADPH氧化成NADP。因此,由于NADPH氧化为NADP,吸光度(340 nm)降低,GPX活性被分光光度法监测。阳性对照采用标准的GPx样硒化有机物依布硒啉。
如图9所示,大部分化合物表现出中等至良好的gpx类活性。对称二硒醚类化合物在本实验中比它们的硒醌类似物表现出更高的活性,这从理论上是可以预料的。此外,化合物9、12、14、19和21的活性更强,是GPx类依布硒啉的5倍。
3.结论
合成了含四唑基的新型对称有机二烯醚类化合物和含硒醌类化合物,采用叠氮- ugi /SN一步反应,产率高。利用三维相似性研究来预测化合物的抗氧化活性,结果显示其静电模式和形状与参考化合物相似,表明其具有类似的GPx模拟活性,这一结果进一步得到了生物筛选的验证。此外,对哺乳动物TrxR1酶的对接研究表明,化合物21、22、23具有良好的应用前景和将硒原子导向Cys 498进行相互作用的结合模式;因此它们可以作为TrxR抑制剂。
此外,用标准MTT法评估化合物对HepG2和MCF-7细胞系的细胞毒性,并与它们对正常的WI-38的细胞毒性进行比较。一般来说,二硒类化合物被发现比硒醌类更具有细胞毒性,并且在HepG2中比在MCF-7细胞中更明显。此外,通过比较HepG2与正常WI-38细胞的细胞毒性,我们发现了明显的选择性。有趣的是,化合物9和18的选择性指数分别为ca. 32和11。此外,化合物6、11、14和20对模型细胞系的治疗指数高达3,而这些治疗指数又都高于基准药物5-FU。在这一点上,一些化合物表现出优先的细胞毒性,这一点在HepG2中与MCF-7细胞比较明显。此外,通过比较正常WI-38细胞和HepG2细胞的细胞毒性,可以观察到明显的选择性模式。
尽管看起来,与一些标准药物相比,这些化合物没有提供纳米摩尔活性,但有足够的证据表明其选择性和未知(因而可能是新的)作用模式的前景值得进一步研究。在这一背景下,我们充分意识到,一个清晰的定量构效关系将需要大量多样的化合物,包括硫,甚至可能需要碲类似物来完成,以确定有机硒的特定作用,并筛选具有改进的活性和选择性的化合物。
如预期的那样,二硒化物显示出优越的GPx催化活性,其中9、12、14、19和21的活性(高达5倍)高于依布硒啉。此外,四唑硒醌20和21在DPPH测定中显示出最高的自由基清除活性,这也符合我们以前的报告,确定醌是有效抗氧化剂在低浓度时。此外,用博来霉素DNA损伤试验评估的合成化合物的亲氧化活性显示化合物14, 19和22能够比其他研究的化合物(例如,6, 9, 10、13和16)诱导显著的DNA降解。
总的来说,硒基醌类化合物是最有前途的候选化合物,因此被选作进一步深入研究分子机制。化合物18、21、22和23对HepG2细胞的治疗显示,与未治疗的细胞相比,Bcl-2和Ki-67的表达水平和caspase-8的激活显著下调。重要的是,23显示出最高的调节活性。这些结果支持了哺乳动物TrxR1的分子对接研究,该研究预测化合物23、21和22的最佳结合能大于18。TrxR1抑制增加caspases水平,从而诱导细胞凋亡。
在这一点上,应当指出,这里提出的结果是初步的,今后需要更深入的研究。因此,有必要进行更深入的研究和额外的实验,以研究确切的作用方式并确定可能的细胞内靶点(如特定的细胞器或细胞膜)。此外,二芳基二硒醚类药物支架的优化设计是提高其药动学和药动学性能的关键。这为进一步开展涉及合成、生物有机和药物化学、细胞生物学和药理学的多学科研究开辟了广阔的空间,以便开发一种应用有机硒化合物治疗癌症的策略。
本文由福山生物整理翻译,转载请注明出处。