






最新资讯



-
电话: 400-113-6988 邮箱: dongfangxicao@163.com 地址: 深圳市南山区粤海街道高新中三道9号环球数码大厦19楼
↓ 微信扫码识别关注我们 ↓ 
硒的表观遗传效应及其对健康的影响
发表于:2019-02-19 作者:admin 来源:本站 点击量:12704
摘要:
表观遗传标记的改变与正常发育和细胞分化以及常见慢性疾病的进展有关。这些标记的可塑性为疾病治疗和预防策略提供了潜力。 已显示大量和微量营养素通过对表观基因组的影响部分地调节疾病风险。必需的微量营养素硒通过硒蛋白和一系列生物活性膳食硒化合物及其代谢产物影响人类健康结果,例如癌症,心血管和自身免疫疾病。该综述提供了关于膳食和合成硒化合物的表观遗传效应的当前文献的评估,其包括表观遗传信息的标记和编辑器的调节以及对单碳代谢的干扰,其提供用于DNA甲基化的甲基供体。讨论了硒 - 表观基因组相互作用对人类健康的相关性,并且我们还指出了未来的研究将有助于更深入地了解硒引起的表观遗传效应。
引言
表观遗传学描述了在不改变基因组DNA序列的情况下调节基因表达的有丝分裂的稳定的染色质机制。这些机制包括DNA的修饰[胞嘧啶甲基化为5-甲基胞嘧啶(5mC)和5mC氧化产物]和组蛋白(乙酰化,甲基化等),其干扰染色体的包装和反式作用因子的结合。DNA / RNA测序技术的最新进展使得有可能在全基因组水平上研究表观遗传标记,从而深入了解所谓的表观基因组。多中心联盟,即DEEP(Deutsches Epigenom-Programm,德国表观基因组计划,www.deutsches-epigenom-programm.de)和IHEC(国际人类表观基因组联盟,www.ihec-epigenomes.org)目前正在破译高分辨率的健康和患病组织/细胞类型的表观基因组图谱,以获得标准的和疾病特异性表观基因组图谱。根据目前的知识,表观基因组在整个生物体的整个生命过程中,在细胞分化过程中以及对多种外部刺激的反应中都表现出可塑性。表观基因组的变化也与癌症和其他复杂疾病的发病和进展有关,如自身免疫性疾病、炎症性肠病、2型糖尿病和心血管疾病。大多数这些关联的因果关系尚不清楚,但考虑到表观遗传标记的主要可变性——与细胞中基本稳定的DNA结构形成对比——以表观基因组为靶点可能在疾病治疗和预防方面提供一个有前途的策略。上述常见疾病的风险和进展的主要决定因素除了遗传易感性外,还有生活方式和饮食等环境因素。已经发现,饮食模式、特定(微)营养素和次生植物化合物会改变表观遗传标记,越来越多的证据表明,食物成分对健康结果的调节(至少部分)是由表观遗传效应介导的。虽然常量营养素(如实验用的高脂肪、高蛋白或卡路里限制饮食)也被证明可以改变表观遗传标记,但由于常量营养素引起的多重混杂效应和成分的变化,很难对这些观察结果作出机理解释。因此,大多数研究已经评估了对特定微量营养素和次生植物化合物干预响应的表观遗传效应。在这方面,硒(Se)是存在于一系列生物活性化合物中的必需微量元素,是特别令人感兴趣的微量营养素。在使用细胞系统和动物的研究中,以及在有限数量的人类研究中,已经发现它可以修改表观遗传标记。Se对维持最佳健康的重要性是基于2种主要硒种群的生物学功能:硒蛋白家族成员,由人类25种基因编码,含有共翻译插入的硒代半胱氨酸,其次,包含在饮食中的非硒蛋白池中的低分子量硒化合物或来自硒代谢。充分表征的硒蛋白,例如谷胱甘肽过氧化物酶(GPx),硫氧还蛋白还原酶(TrxR)和碘甲状腺原氨酸脱碘酶(DIO)是氧化还原酶,并且充当抗氧化酶,氧化还原敏感信号传导途径和甲状腺激素代谢的调节剂。其他硒蛋白促进硒转运(硒蛋白P),硒蛋白生物合成(硒磷酸合成酶2),并参与维持内质网稳态(例如,硒蛋白S,K和15-kDa硒蛋白)。硒蛋白对小鼠发育和健康的重要性已经在具有单一或完全硒蛋白消耗的转基因小鼠中得到证实。此外,具有遗传上受损的硒蛋白生物合成的人患有严重的多系统疾病。 已知小鼠和人硒补充研究的生物学结果(例如,疾病发生率,对转录组,表观基因组和硒状态的影响(及其中的参考文献),不仅受Se剂量的影响,而且还受其化学形式和补充前Se状态的影响。因此,我们将简要介绍硒的需求和膳食硒化合物的代谢。此后,我们总结了目前对天然存在的和合成的硒化合物对表观遗传标记和编辑的影响的知识,并讨论了它们对健康和疾病的可能相关性。目的是研究Se -表观基因组相互作用的生化基础,为此,我们将重点研究Se化合物对表观遗传机制(如DNA甲基转移酶、组蛋白修饰酶、单碳代谢)的直接和间接影响。目前的知识还不能完全回答这些问题,因此我们指出哪些研究可能有助于拓宽我们对未来se -表观基因组相互作用的理解。
硒的需求量和膳食硒化合物的代谢
在西方国家,显性硒缺乏症相对较少。与硒缺乏相关的一种临床症状是克山病,它发生在中国的一个省份,受影响的人每日总硒摄入量≤15μg。据世界卫生组织(World Health Organization)的定义,可容忍的硒摄入量上限为每日400μg,大多数卫生机构建议每日摄入的硒量在55至70μg之间。这些建议通常基于总硒摄入量来优化血浆中GPx3和SeP的活性/表达,这是2种常用的硒状态生物标志物。实现血浆GPx3活性的优化在40-47μg Se /天,而SeP需要~105μg Se /天。与SeP优化(124μg Se/l)一致的血浆硒浓度处于与降低死亡率风险和预防几种癌症相关的范围内。超过这些硒水平的补充似乎不会带来额外的益处,但可能会增加2型糖尿病的风险。主要的膳食硒化合物是氨基酸硒代蛋氨酸(SelMet),硒代半胱氨酸,Se-甲基硒代半胱氨酸,以及阴离子亚硒酸盐和硒酸盐(用于列出硒化合物和食品中的数量)。Se化合物通过不同的途径(如图1所示)代谢成硒化氢。硒化氢被甲基化为排泄形式(二甲基硒,三甲基硒,Se-甲基-N-乙酰基硒基己糖胺)或磷酸化为硒磷酸盐,用作氨基酸硒代半胱氨酸(Sec)的前体,其从Sec特异性tRNA(Sec- tRNA [Ser] Sec)共翻译插入进入硒蛋白。亚硒酸盐通过谷胱甘肽化还原为硒(/二)谷胱甘肽或通过谷氧还蛋白直接还原为硒化物。SelMet的代谢通过转硫途径发生 - 由相同的酶催化将甲硫氨酸转化为半胱氨酸至Sec,其通过硒代半胱氨酸β-裂解酶(SBL)转化为硒化物和丙氨酸。在类似的反应中,SBL从Se-甲基硒代半胱氨酸产生甲基硒醇。甲基硒基氨基酸Se-甲基硒代半胱氨酸和SelMet可以分别通过谷氨酰胺转氨酶K和L-氨基酸氧化酶转氨基转化为β-甲基硒基丙酮酸和α-酮基-γ-甲基硒基丁酸酯(参见Se代谢参考文献见24)。
表观遗传标记的改变与正常发育和细胞分化以及常见慢性疾病的进展有关。这些标记的可塑性为疾病治疗和预防策略提供了潜力。 已显示大量和微量营养素通过对表观基因组的影响部分地调节疾病风险。必需的微量营养素硒通过硒蛋白和一系列生物活性膳食硒化合物及其代谢产物影响人类健康结果,例如癌症,心血管和自身免疫疾病。该综述提供了关于膳食和合成硒化合物的表观遗传效应的当前文献的评估,其包括表观遗传信息的标记和编辑器的调节以及对单碳代谢的干扰,其提供用于DNA甲基化的甲基供体。讨论了硒 - 表观基因组相互作用对人类健康的相关性,并且我们还指出了未来的研究将有助于更深入地了解硒引起的表观遗传效应。
引言
表观遗传学描述了在不改变基因组DNA序列的情况下调节基因表达的有丝分裂的稳定的染色质机制。这些机制包括DNA的修饰[胞嘧啶甲基化为5-甲基胞嘧啶(5mC)和5mC氧化产物]和组蛋白(乙酰化,甲基化等),其干扰染色体的包装和反式作用因子的结合。DNA / RNA测序技术的最新进展使得有可能在全基因组水平上研究表观遗传标记,从而深入了解所谓的表观基因组。多中心联盟,即DEEP(Deutsches Epigenom-Programm,德国表观基因组计划,www.deutsches-epigenom-programm.de)和IHEC(国际人类表观基因组联盟,www.ihec-epigenomes.org)目前正在破译高分辨率的健康和患病组织/细胞类型的表观基因组图谱,以获得标准的和疾病特异性表观基因组图谱。根据目前的知识,表观基因组在整个生物体的整个生命过程中,在细胞分化过程中以及对多种外部刺激的反应中都表现出可塑性。表观基因组的变化也与癌症和其他复杂疾病的发病和进展有关,如自身免疫性疾病、炎症性肠病、2型糖尿病和心血管疾病。大多数这些关联的因果关系尚不清楚,但考虑到表观遗传标记的主要可变性——与细胞中基本稳定的DNA结构形成对比——以表观基因组为靶点可能在疾病治疗和预防方面提供一个有前途的策略。上述常见疾病的风险和进展的主要决定因素除了遗传易感性外,还有生活方式和饮食等环境因素。已经发现,饮食模式、特定(微)营养素和次生植物化合物会改变表观遗传标记,越来越多的证据表明,食物成分对健康结果的调节(至少部分)是由表观遗传效应介导的。虽然常量营养素(如实验用的高脂肪、高蛋白或卡路里限制饮食)也被证明可以改变表观遗传标记,但由于常量营养素引起的多重混杂效应和成分的变化,很难对这些观察结果作出机理解释。因此,大多数研究已经评估了对特定微量营养素和次生植物化合物干预响应的表观遗传效应。在这方面,硒(Se)是存在于一系列生物活性化合物中的必需微量元素,是特别令人感兴趣的微量营养素。在使用细胞系统和动物的研究中,以及在有限数量的人类研究中,已经发现它可以修改表观遗传标记。Se对维持最佳健康的重要性是基于2种主要硒种群的生物学功能:硒蛋白家族成员,由人类25种基因编码,含有共翻译插入的硒代半胱氨酸,其次,包含在饮食中的非硒蛋白池中的低分子量硒化合物或来自硒代谢。充分表征的硒蛋白,例如谷胱甘肽过氧化物酶(GPx),硫氧还蛋白还原酶(TrxR)和碘甲状腺原氨酸脱碘酶(DIO)是氧化还原酶,并且充当抗氧化酶,氧化还原敏感信号传导途径和甲状腺激素代谢的调节剂。其他硒蛋白促进硒转运(硒蛋白P),硒蛋白生物合成(硒磷酸合成酶2),并参与维持内质网稳态(例如,硒蛋白S,K和15-kDa硒蛋白)。硒蛋白对小鼠发育和健康的重要性已经在具有单一或完全硒蛋白消耗的转基因小鼠中得到证实。此外,具有遗传上受损的硒蛋白生物合成的人患有严重的多系统疾病。 已知小鼠和人硒补充研究的生物学结果(例如,疾病发生率,对转录组,表观基因组和硒状态的影响(及其中的参考文献),不仅受Se剂量的影响,而且还受其化学形式和补充前Se状态的影响。因此,我们将简要介绍硒的需求和膳食硒化合物的代谢。此后,我们总结了目前对天然存在的和合成的硒化合物对表观遗传标记和编辑的影响的知识,并讨论了它们对健康和疾病的可能相关性。目的是研究Se -表观基因组相互作用的生化基础,为此,我们将重点研究Se化合物对表观遗传机制(如DNA甲基转移酶、组蛋白修饰酶、单碳代谢)的直接和间接影响。目前的知识还不能完全回答这些问题,因此我们指出哪些研究可能有助于拓宽我们对未来se -表观基因组相互作用的理解。
硒的需求量和膳食硒化合物的代谢
在西方国家,显性硒缺乏症相对较少。与硒缺乏相关的一种临床症状是克山病,它发生在中国的一个省份,受影响的人每日总硒摄入量≤15μg。据世界卫生组织(World Health Organization)的定义,可容忍的硒摄入量上限为每日400μg,大多数卫生机构建议每日摄入的硒量在55至70μg之间。这些建议通常基于总硒摄入量来优化血浆中GPx3和SeP的活性/表达,这是2种常用的硒状态生物标志物。实现血浆GPx3活性的优化在40-47μg Se /天,而SeP需要~105μg Se /天。与SeP优化(124μg Se/l)一致的血浆硒浓度处于与降低死亡率风险和预防几种癌症相关的范围内。超过这些硒水平的补充似乎不会带来额外的益处,但可能会增加2型糖尿病的风险。主要的膳食硒化合物是氨基酸硒代蛋氨酸(SelMet),硒代半胱氨酸,Se-甲基硒代半胱氨酸,以及阴离子亚硒酸盐和硒酸盐(用于列出硒化合物和食品中的数量)。Se化合物通过不同的途径(如图1所示)代谢成硒化氢。硒化氢被甲基化为排泄形式(二甲基硒,三甲基硒,Se-甲基-N-乙酰基硒基己糖胺)或磷酸化为硒磷酸盐,用作氨基酸硒代半胱氨酸(Sec)的前体,其从Sec特异性tRNA(Sec- tRNA [Ser] Sec)共翻译插入进入硒蛋白。亚硒酸盐通过谷胱甘肽化还原为硒(/二)谷胱甘肽或通过谷氧还蛋白直接还原为硒化物。SelMet的代谢通过转硫途径发生 - 由相同的酶催化将甲硫氨酸转化为半胱氨酸至Sec,其通过硒代半胱氨酸β-裂解酶(SBL)转化为硒化物和丙氨酸。在类似的反应中,SBL从Se-甲基硒代半胱氨酸产生甲基硒醇。甲基硒基氨基酸Se-甲基硒代半胱氨酸和SelMet可以分别通过谷氨酰胺转氨酶K和L-氨基酸氧化酶转氨基转化为β-甲基硒基丙酮酸和α-酮基-γ-甲基硒基丁酸酯(参见Se代谢参考文献见24)。

图1. 膳食硒化合物的代谢。 主要的有机和无机硒化合物通过转硫化,转氨作用和硫氧还蛋白还原酶,谷胱甘肽还原酶和谷氧还蛋白的还原代谢。 标有绿色背景的是参与的酶。 详情请见文字。 GTK =谷氨酰胺转氨酶K; AAO= L-氨基酸氧化酶; GR=谷胱甘肽还原酶; CGL=胱硫醚γ-裂解酶; CBS =胱硫醚β-合成酶; SBL =硒代半胱氨酸β-裂解酶。
硒对DNA和组蛋白表观遗传修饰的影响
硒对DNA甲基化的影响
基因组DNA中胞嘧啶的甲基化是高等生物中最常见且可能研究最多的表观遗传修饰。甲基在DNA甲基转移酶(DNMT1,DNMT2,DNMT3A,DNMT3B和DNMT3L)催化的反应中转移,从供体底物S-腺苷甲硫氨酸(SAM)到胞嘧啶的5-碳位置 - 通常当它与鸟苷结合时在3’-,得到5-甲基胞嘧啶(5mC)。另一方面,DNA去甲基化不是直接催化的,而是由DNA复制偶联稀释产生的,其中5mC或5mC氧化产物[5-羟甲基胞嘧啶(5hmC),5-甲酰基胞嘧啶(5fC)和5-羧基胞嘧啶 (5 caC)]不会被复制到新的DNA链,或者通过碱基切除修复(BER)或核苷酸切除修复(NER)将5 mC(或衍生物)与短的周围核苷酸段一起替换。因此,营养物质对DNA甲基化的干扰主要是通过(i) DNMT活性/与附属因子的相互作用、(ii) SAM可用性和(iii)去甲基化过程发生的。许多研究报道了Se状态或补充对总DNA和基因特异性DNA甲基化以及DNMT的表达或活性的影响(列于表1)。Arai等人用母体血清中发现的生理,无毒Se剂量孵育小鼠胚胎干细胞。Se引起细胞异染色质状态的可逆改变,并且还改变了在胎儿发育中起作用的个体基因的DNA甲基化状态,包括Hnf4a(肝细胞核因子4a),Aebp2(AE结合蛋白2),Prickle2(刺猬同源物2), 和Rnd2(Rho家族GTP酶2),不损害细胞形成胚胎体的潜力。这些结果意味着通过对基因特异性甲基化的影响,Se与组织特异性分化之间存在有趣的联系,因为众所周知Se是体外肝细胞分化所必需的,转录因子HNF4a是该过程的关键调节因子。Arai等人观察到的染色质结构的变化可能是由Se补充细胞和缺陷细胞的总DNA甲基化差异引起的。使用啮齿动物和细胞系的研究确实表明饮食中Se摄入水平影响总DNA甲基化。啮齿动物研究给出了关于补充硒对总DNA甲基化增加或减少的不一致结果,尽管使用了可比较的硒饮食(表1):硒缺乏导致大鼠肝脏和结肠中DNA甲基化较少,与Zeng等人的研究相反,其中喂食超营养的大鼠与足够和缺乏的Se饮食的相比,肝脏和结肠中的DNA甲基化水平较高。Zeng等人指出,所用动物菌株和基础饮食含量的差异可能是硒效应的改良剂。此外,采用不同的技术对总DNA甲基化进行评估:采用[3H -甲基]-SAM/SssI甲基化酶和分离的DNA 进行体外甲基群接受试验,一个5 mC 酶联免疫吸附剂测定(ELISA),用高效液相色谱(HPLC)检测酶解DNA中的5mC单磷酸腺苷。体外研究的相关数据仅限于一篇论文,显示LNCaP前列腺肿瘤细胞经1.5 μM亚硒酸盐处理7 d后,5 mC免疫反应活性下降约50%。可以得出的结论是,亚硒酸盐和硒代蛋氨酸(SelMet)对总DNA甲基化的影响可能被菌株特异性效应所掩盖,而且它们还受到营养环境(如高脂肪饮食)的影响。该主题已在人类研究(N =287)中阐明,该研究发现血浆Se与白细胞中的总DNA甲基化呈显著负相关。除了对整体甲基化的影响外,硒还被证明能在个体基因的区域和特定的CpG位点诱导不同的甲基化。Xiang等人的研究发现,编码II相解毒酶GSTP1(π-型谷胱甘肽S-转移酶)和肿瘤抑制因子APC(腺瘤性息肉病)和CSR1(细胞应激反应1)的基因由于其启动子的高甲基化而在前列腺肿瘤中经常被沉默,在亚硝酸盐处理后在LNCaP细胞中去甲基化并重新表达。类似地,不同来源的Se(100μM SelMet)引起启动子去甲基化和GSTP1的重新表达。硒化合物通常以10nM至100μM的浓度用于体外研究。Se的生长抑制和毒性作用取决于其化学形式和细胞类型。 虽然Se与蛋白质(如血浆中)或氨基酸(例如SelMet)结合时具有低毒性,但许多细胞系不能耐受1μM剂量的亚硒酸盐或甲基硒酸。考虑到人血浆中Se的生理浓度范围(~0.4-2.5μM),> 5μM的Se剂量是超生理学的,不适用于补充试验。在前列腺癌的预防中使用硒是非常有前途的,这是基于对前列腺癌风险和硒状态反向关联的观察,以及支持硒蛋白缺陷小鼠中加速前列腺癌发生的发现。在这种情况下,可能会出现一种概念,认为Se是一种通过靶向肿瘤抑制基因来抵抗癌症进展的表观遗传药物,如Xiang等人所暗示的那样,但当然需要进行体内研究以加强和扩展其他相关基因,这些基因也可能以不同形式和不同的癌变阶段被Se靶向。发现von-Hippel-Lindau(VHL综合征)基因启动子的甲基化对Caco-2细胞中的Se(250nM Se-甲基硒代半胱氨酸(SeMSC))起反应; 体外培养的VHL启动子甲基化被SeMSC降低,这与Caco-2和2 μg Se(SeMSC)喂养的大鼠VHL表达水平升高有关。VHL经常在肾细胞癌中下调和突变,并且还发现在结肠直肠癌发生期间失调,其中Se基于流行病学和动物研究被归因于保护功能。一项针对人类的研究评估了与硒状态相关的健康直肠粘膜标本(84名男性,101名女性)中结肠直肠癌相关基因的甲基化状态。 发现WIF1(wnt抑制因子1)甲基化和血浆Se浓度的关联。有趣的是,硒的状态也与其他基因和反转录转座子的甲基化有关,包括LINE1(长散在的核苷酸元件1),PCA1(阳离子转运P型ATP酶),SFRP1 / 2(分泌的卷曲相关蛋白1/2), 和APC,这是以性别特异性的方式发生的。没有分析这些基因的差异甲基化是否与其表达水平的差异相关。EPIC(欧洲癌症和营养队列前瞻性调查)的研究揭示了Se状态与结直肠癌风险呈负相关关系,这种关系在女性中比男性强。硒蛋白表达水平和硒状态生物标志物对男性和女性补充硒的不同反应也可以明显看出硒的性别特异性效应(总结见参考文献38); Tapp等人的研究表明,Se效应的这种性别特异性延伸到癌症相关基因的表观遗传标记,但这些发现与疾病病因学的相关性值得进一步研究。最近的一项研究将炎症相关基因TLR2(toll样受体2)和ICAM1(细胞间粘附分子1)鉴定为Se依赖性表观遗传调控的新靶点,并提出了一种机制,即Se改变GADD45的表达(生长停滞和DNA损伤诱导基因,α)和DNMT1,导致TLR2和ICAM1的表观遗传沉默,并将其与由长期严重缺乏的硒摄入引起的众所周知的病症联系起来:克山病(KD)。KD是一种病毒性心肌炎,心肌坏死病灶于1935年首次在中国东北的克山县发现,在中国缺硒地区流行。结果发现,Se缺乏是KD的致病因素,Se作为补充剂后发病率显著降低。Yang等人使用甲基化DNA-IP比较了来自KD患者和健康对照组的外周血中的DNA甲基化,并随后通过Roche-Nimblegen HG18 CpG启动子阵列分析了富集的DNA。甲基化组谱显示在数千个差异甲基化区域(DMR)的差异,其通过甲基化特异性PCR证实了TLR2和ICAM1启动子。此外,两种基因的表达水平与启动子甲基化程度和血清Se浓度呈负相关。用喂食含有0,0.1和2.0mg / kg亚硒酸盐的饮食的大鼠获得了类似的结果,而与人类受试者相比,在心肌组织和分离的新生儿心肌细胞中测量了Tlr2和Icam1基因的甲基化/表达。虽然与Se缺陷细胞相比,在用1.5μM亚硒酸盐处理的心肌细胞中观察到Dnmt1蛋白表达水平的显著降低,但这不太可能并且预期是Tlr2和Icam1启动子甲基化增加的原因。相反,作者认为这是由于Se处理的心肌细胞中Gadd45a mRNA和蛋白质表达水平的降低。GADD45A通过与BER-和NER-执行蛋白的相互作用与特定基因组位点,例如在RARβ2(视黄酸受体β)的基因中的去甲基化相关联。但是,正如Yang等人暗示的那样,GADD45A作为Se依赖性调节(位点特异性)DNA甲基化的介质的作用仍然是推测,直到通过使用例如GADD45A基因沉默和染色质免疫沉淀的其他研究证实。此外,许多研究报道,Se通过增强的p53结合活性增强DNA损伤修复能力(总结于42),部分地与大鼠心肌细胞获得的结果相反,通过GPx-1-与MCF7细胞中GADD45的表达增加相关或其与DNA修复酶如AP内切核酸酶1的相互作用相关。然而,Se对DNA修复酶的这些作用是否确实促进DNA去甲基化尚不确定。
发现WIF1(wnt抑制因子1)甲基化和血浆Se浓度的关联。有趣的是,硒的状态也与其他基因和反转录转座子的甲基化有关,包括LINE1(长散在的核苷酸元件1),PCA1(阳离子转运P型ATP酶),SFRP1 / 2(分泌的卷曲相关蛋白1/2), 和APC,这是以性别特异性的方式发生的。没有分析这些基因的差异甲基化是否与其表达水平的差异相关。EPIC(欧洲癌症和营养队列前瞻性调查)的研究揭示了Se状态与结直肠癌风险呈负相关关系,这种关系在女性中比男性强。硒蛋白表达水平和硒状态生物标志物对男性和女性补充硒的不同反应也可以明显看出硒的性别特异性效应(总结见参考文献38); Tapp等人的研究表明,Se效应的这种性别特异性延伸到癌症相关基因的表观遗传标记,但这些发现与疾病病因学的相关性值得进一步研究。最近的一项研究将炎症相关基因TLR2(toll样受体2)和ICAM1(细胞间粘附分子1)鉴定为Se依赖性表观遗传调控的新靶点,并提出了一种机制,即Se改变GADD45的表达(生长停滞和DNA损伤诱导基因,α)和DNMT1,导致TLR2和ICAM1的表观遗传沉默,并将其与由长期严重缺乏的硒摄入引起的众所周知的病症联系起来:克山病(KD)。KD是一种病毒性心肌炎,心肌坏死病灶于1935年首次在中国东北的克山县发现,在中国缺硒地区流行。结果发现,Se缺乏是KD的致病因素,Se作为补充剂后发病率显著降低。Yang等人使用甲基化DNA-IP比较了来自KD患者和健康对照组的外周血中的DNA甲基化,并随后通过Roche-Nimblegen HG18 CpG启动子阵列分析了富集的DNA。甲基化组谱显示在数千个差异甲基化区域(DMR)的差异,其通过甲基化特异性PCR证实了TLR2和ICAM1启动子。此外,两种基因的表达水平与启动子甲基化程度和血清Se浓度呈负相关。用喂食含有0,0.1和2.0mg / kg亚硒酸盐的饮食的大鼠获得了类似的结果,而与人类受试者相比,在心肌组织和分离的新生儿心肌细胞中测量了Tlr2和Icam1基因的甲基化/表达。虽然与Se缺陷细胞相比,在用1.5μM亚硒酸盐处理的心肌细胞中观察到Dnmt1蛋白表达水平的显著降低,但这不太可能并且预期是Tlr2和Icam1启动子甲基化增加的原因。相反,作者认为这是由于Se处理的心肌细胞中Gadd45a mRNA和蛋白质表达水平的降低。GADD45A通过与BER-和NER-执行蛋白的相互作用与特定基因组位点,例如在RARβ2(视黄酸受体β)的基因中的去甲基化相关联。但是,正如Yang等人暗示的那样,GADD45A作为Se依赖性调节(位点特异性)DNA甲基化的介质的作用仍然是推测,直到通过使用例如GADD45A基因沉默和染色质免疫沉淀的其他研究证实。此外,许多研究报道,Se通过增强的p53结合活性增强DNA损伤修复能力(总结于42),部分地与大鼠心肌细胞获得的结果相反,通过GPx-1-与MCF7细胞中GADD45的表达增加相关或其与DNA修复酶如AP内切核酸酶1的相互作用相关。然而,Se对DNA修复酶的这些作用是否确实促进DNA去甲基化尚不确定。
基因组DNA中胞嘧啶的甲基化是高等生物中最常见且可能研究最多的表观遗传修饰。甲基在DNA甲基转移酶(DNMT1,DNMT2,DNMT3A,DNMT3B和DNMT3L)催化的反应中转移,从供体底物S-腺苷甲硫氨酸(SAM)到胞嘧啶的5-碳位置 - 通常当它与鸟苷结合时在3’-,得到5-甲基胞嘧啶(5mC)。另一方面,DNA去甲基化不是直接催化的,而是由DNA复制偶联稀释产生的,其中5mC或5mC氧化产物[5-羟甲基胞嘧啶(5hmC),5-甲酰基胞嘧啶(5fC)和5-羧基胞嘧啶 (5 caC)]不会被复制到新的DNA链,或者通过碱基切除修复(BER)或核苷酸切除修复(NER)将5 mC(或衍生物)与短的周围核苷酸段一起替换。因此,营养物质对DNA甲基化的干扰主要是通过(i) DNMT活性/与附属因子的相互作用、(ii) SAM可用性和(iii)去甲基化过程发生的。许多研究报道了Se状态或补充对总DNA和基因特异性DNA甲基化以及DNMT的表达或活性的影响(列于表1)。Arai等人用母体血清中发现的生理,无毒Se剂量孵育小鼠胚胎干细胞。Se引起细胞异染色质状态的可逆改变,并且还改变了在胎儿发育中起作用的个体基因的DNA甲基化状态,包括Hnf4a(肝细胞核因子4a),Aebp2(AE结合蛋白2),Prickle2(刺猬同源物2), 和Rnd2(Rho家族GTP酶2),不损害细胞形成胚胎体的潜力。这些结果意味着通过对基因特异性甲基化的影响,Se与组织特异性分化之间存在有趣的联系,因为众所周知Se是体外肝细胞分化所必需的,转录因子HNF4a是该过程的关键调节因子。Arai等人观察到的染色质结构的变化可能是由Se补充细胞和缺陷细胞的总DNA甲基化差异引起的。使用啮齿动物和细胞系的研究确实表明饮食中Se摄入水平影响总DNA甲基化。啮齿动物研究给出了关于补充硒对总DNA甲基化增加或减少的不一致结果,尽管使用了可比较的硒饮食(表1):硒缺乏导致大鼠肝脏和结肠中DNA甲基化较少,与Zeng等人的研究相反,其中喂食超营养的大鼠与足够和缺乏的Se饮食的相比,肝脏和结肠中的DNA甲基化水平较高。Zeng等人指出,所用动物菌株和基础饮食含量的差异可能是硒效应的改良剂。此外,采用不同的技术对总DNA甲基化进行评估:采用[3H -甲基]-SAM/SssI甲基化酶和分离的DNA 进行体外甲基群接受试验,一个5 mC 酶联免疫吸附剂测定(ELISA),用高效液相色谱(HPLC)检测酶解DNA中的5mC单磷酸腺苷。体外研究的相关数据仅限于一篇论文,显示LNCaP前列腺肿瘤细胞经1.5 μM亚硒酸盐处理7 d后,5 mC免疫反应活性下降约50%。可以得出的结论是,亚硒酸盐和硒代蛋氨酸(SelMet)对总DNA甲基化的影响可能被菌株特异性效应所掩盖,而且它们还受到营养环境(如高脂肪饮食)的影响。该主题已在人类研究(N =287)中阐明,该研究发现血浆Se与白细胞中的总DNA甲基化呈显著负相关。除了对整体甲基化的影响外,硒还被证明能在个体基因的区域和特定的CpG位点诱导不同的甲基化。Xiang等人的研究发现,编码II相解毒酶GSTP1(π-型谷胱甘肽S-转移酶)和肿瘤抑制因子APC(腺瘤性息肉病)和CSR1(细胞应激反应1)的基因由于其启动子的高甲基化而在前列腺肿瘤中经常被沉默,在亚硝酸盐处理后在LNCaP细胞中去甲基化并重新表达。类似地,不同来源的Se(100μM SelMet)引起启动子去甲基化和GSTP1的重新表达。硒化合物通常以10nM至100μM的浓度用于体外研究。Se的生长抑制和毒性作用取决于其化学形式和细胞类型。 虽然Se与蛋白质(如血浆中)或氨基酸(例如SelMet)结合时具有低毒性,但许多细胞系不能耐受1μM剂量的亚硒酸盐或甲基硒酸。考虑到人血浆中Se的生理浓度范围(~0.4-2.5μM),> 5μM的Se剂量是超生理学的,不适用于补充试验。在前列腺癌的预防中使用硒是非常有前途的,这是基于对前列腺癌风险和硒状态反向关联的观察,以及支持硒蛋白缺陷小鼠中加速前列腺癌发生的发现。在这种情况下,可能会出现一种概念,认为Se是一种通过靶向肿瘤抑制基因来抵抗癌症进展的表观遗传药物,如Xiang等人所暗示的那样,但当然需要进行体内研究以加强和扩展其他相关基因,这些基因也可能以不同形式和不同的癌变阶段被Se靶向。发现von-Hippel-Lindau(VHL综合征)基因启动子的甲基化对Caco-2细胞中的Se(250nM Se-甲基硒代半胱氨酸(SeMSC))起反应; 体外培养的VHL启动子甲基化被SeMSC降低,这与Caco-2和2 μg Se(SeMSC)喂养的大鼠VHL表达水平升高有关。VHL经常在肾细胞癌中下调和突变,并且还发现在结肠直肠癌发生期间失调,其中Se基于流行病学和动物研究被归因于保护功能。一项针对人类的研究评估了与硒状态相关的健康直肠粘膜标本(84名男性,101名女性)中结肠直肠癌相关基因的甲基化状态。
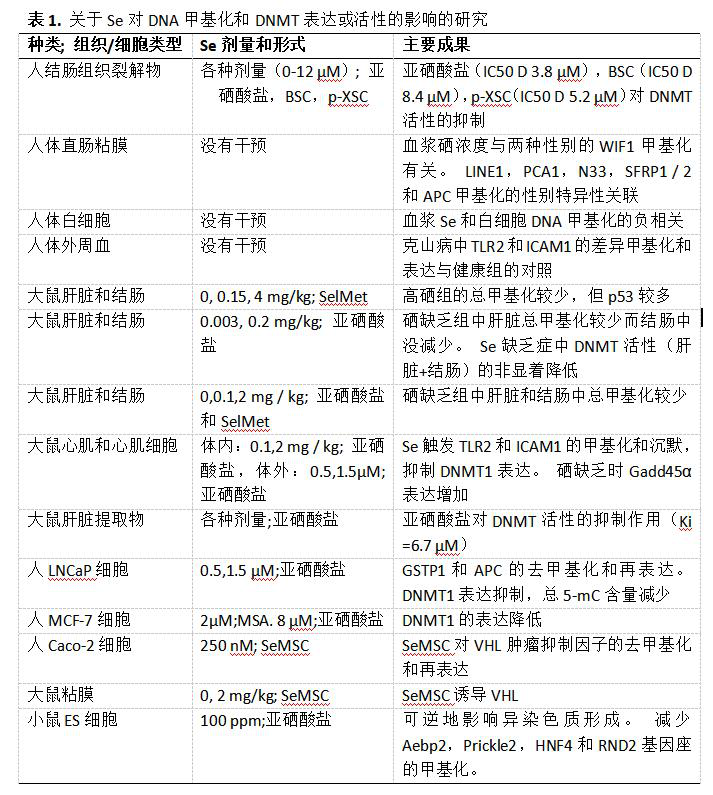
另一种可能诱导差异DNA甲基化的se靶点是DNMT酶;在上述研究中发现,DNMT表达或活性的调节被认为有助于甲基化标记的调节。亚硒酸盐和2种合成硒化合物,苄基硒氰酸盐(BSC)和1,4-亚苯基双(亚甲基)硒氰酸盐(p-XSC)已被证明可抑制人结肠癌核提取物中的DNMT活性。3种化合物的IC 50值计算为3.8μM(亚硒酸盐),8.4μM(BSC)和5.2μM(p-XSC),实验的设置暗示化合物以其非代谢形式起作用。在体外试验中,由大鼠肝脏制备的DNMT也在6.7μM的Ki下被亚硒酸盐抑制,并且与对照动物组相比,从Se补充分离时酶活性较低。在体内,在喂食Se-足够与缺乏的饮食(0.2和0mg Se / kg饮食作为亚硒酸钠)的大鼠的肝脏和结肠中观察到DNMT活性的非显著降低。类似地,在大鼠心肌细胞和LNCaP细胞中通过1.5μM亚硒酸钠和在MCF-7细胞中通过8μM亚硒酸钠和2μM MSA,减少DNMT1蛋白表达。体外人和动物研究共同表明Se与总DNA甲基化和DNMT活性呈负相关。
硒对组蛋白乙酰化的影响
组蛋白蛋白携带多种翻译后修饰(如甲基化和乙酰化),由组蛋白甲基转移酶、组蛋白乙酰转移酶(HATs)等在确定的氨基酸位置添加。由IHEC联盟研究的常见组蛋白标记是组蛋白3的赖氨酸残基4,9,27和36处的甲基化和乙酰化。组蛋白标记的高度多样化编码控制组蛋白与DNA的结合、DNA与反转录因子的相互作用以及最终的基因表达。在疾病的发病和进展中发现异常的组蛋白密码,因此成为治疗目标。硒和其他微量营养素已被证明可诱导或与组蛋白标记的变化相关,从而可能影响健康结果。营养物质对组蛋白标记的干扰主要是通过调节组蛋白修饰酶活性/表达和对底物有效性的干扰发生的。鉴于标记和参与酶的种类繁多,情况甚至比DNA甲基化更复杂; 此外,DNA甲基化和组蛋白标记之间存在交叉链,它们一起构成了一个复杂的表观遗传调控网络。从临床角度来看,对组蛋白脱乙酰酶(HDACs)特别感兴趣,因为它们的异常功能和/或表达与癌症和一些神经和免疫疾病有关。已经开发了许多合成的HDAC抑制剂,并且目前正在临床试验中进行测试。一些天然存在的饮食因素或代谢物如丁酸盐,多酚和硒也已被证明可作为HDAC抑制剂。报告通过膳食和合成Se化合物调节组蛋白标记和编辑酶(HDACs和HATs)的研究列于表2中并在下面讨论。
表2.通过膳食和合成硒化合物调节组蛋白修饰
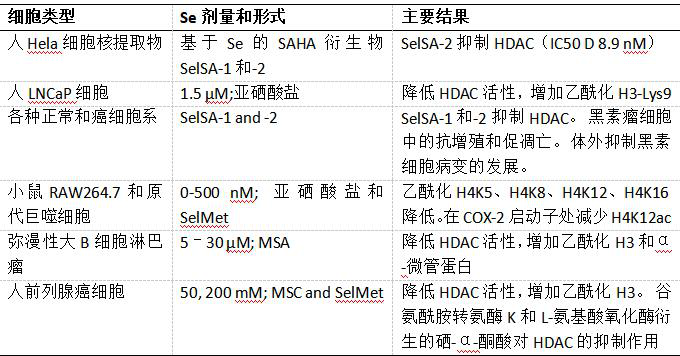

图2. 含有Se的HDAC活性抑制剂。 硒代谢物(β-甲基硒丙酮酸和α-酮-γ甲基硒基丁酸酯)和合成硒化合物(B(PCP)-2Se [双(5-苯基氨基甲酰基戊基)二硒醚],SelSA-1和PCP-SeCN(5-苯基氨基甲酰基戊基硒氰化物),SelSA-2)的结构式具有HDAC抑制活性。
用1.5μM亚硒酸盐处理LNCaP细胞7天显着降低HDAC活性,同时伴随轻度降低的HDAC3和未改变的HDAC4和HDAC5蛋白水平。与降低的HDAC活性一致,作者发现,在Se处理后,总的和GSTP1启动子结合的H3K9ac水平(一个 抑制标记) 增加,H3K9me3(一个活化标记)降低。其他研究证实,膳食和合成硒化合物可抑制HDAC活性。Kassam等人应用甲基硒酸(MSA; 5-30μM)扩散大B细胞淋巴瘤(DLBCL)细胞系并观察到HDAC活性降低和HDAC靶向乙酰化组蛋白H3和乙酰化α-微管蛋白的总水平增加。这些效应需要MSA的细胞代谢,但HDAC失活的代谢物尚未被确认。Lee等人发现来自膳食硒化合物的硒代α-酮酸代谢物充当HDACs的抑制剂。在他们的研究中,SeMSC和SelMet在谷氨酰胺转氨酶K(GTK)和L-氨基酸氧化酶催化的转氨作用中反应为β-甲基硒丙酮酸和α-酮-γ - 甲基硒基丁酸,其结构与众所周知的HDAC抑制剂丁酸盐相似(图2)。与SeMSC和SelMet相比,硒代-α-酮酸均抑制无细胞试验中的HDAC活性,并导致前列腺癌细胞中乙酰化组蛋白H3水平的快速升高。在实验性癌发生的研究中观察到的高剂量Se补充的抗癌作用部分归因于HDAC的抑制并且刺激了更有效的含Se的HDAC抑制剂(HDACi)的合成。基于辛二酰苯胺异羟肟酸(SAHA,商业上可用作伏立诺他)的结构,用于治疗晚期T细胞淋巴瘤的HDACi,2个基于Se的衍生物被合成,命名为SelSA-1和SelSA-2(图2)。SelSA-2对HeLa核细胞提取物(主要是同种型HDAC1和HDAC2)的HDAC抑制活性高于SAHA和曲古抑菌素A。两种硒化合物在抑制皮肤重建中的黑素瘤细胞生长和黑素细胞病变发展方面也更有效,但它们作为针对黑素瘤或其他癌症的药物的用途尚未经过测试。最近的一篇论文表明,在用亚硒酸盐(100-500nM)处理的巨噬细胞中,其赖氨酸残基5,8,12和16处的组蛋白H4乙酰化降低。与此同时,促炎性基因肿瘤坏死因子-α(TNF-α))和环氧合酶-2 (COX2)的启动子中H4K12ac和H4K16ac的丰度下降,此前已在se处理的RAW264.7巨噬细胞中表达减少。这些细胞中的HDAC活性不受亚硒酸盐的影响,作者提出亚硒酸盐引发的H4乙酰化水平降低是由于通过硒依赖性和造血前列腺素D合成酶(H-PGDS)抑制p300 HAT活性 - 和 COX介导抗炎Δ-PGJ和15d-PGJ的产生,其可以共价结合p300从而抑制其活性。重要的是,亚硒酸盐没有改变缺乏Sec-tRNA[Ser] Sec 的巨噬细胞中的H4乙酰化水平,表明硒蛋白生物合成是亚硒酸盐诱导的H4乙酰化调节的先决条件。由亚硒酸盐(涉及H-PGDS,COX-2,p300 HAT和p65和H4乙酰化)引发的所提出的级联反应,可能触发从M1转变为巨噬细胞的M2表型。因此,这也有助于理解硒和硒蛋白在炎症性肠病和其他慢性炎症疾病中的抗炎作用。
硒与单碳代谢的相互关系
一碳代谢途径(如图3所示)提供甲基供体S-腺苷甲硫氨酸(SAM), DNMTs和其他甲基化酶作为底物将甲基转移到细胞因子和靶蛋白。因此,单碳代谢与DNA甲基化紧密相关,并且影响一碳代谢的条件,特别是参与酶的辅助因子叶酸,胆碱/甜菜碱和维生素B2,B6和B12的可用性,显示导致差异DNA甲基化。我们简要描述了一碳代谢和相关途径的反应,然后评估了当前的文献关于其硒依赖性调节的发现。如图3所示,甲硫氨酸与SAM反应,在DNMT催化的胞嘧啶或蛋白质甲基化后,SAM转化为S-腺苷高半胱氨酸(SAH)。去除腺苷基团得到同型半胱氨酸(HCys)。血液或血浆中的HCys浓度具有临床重要性,因为它与多种复杂疾病如心血管和神经退行性疾病(及其中的参考文献)有关。HCys的清除作用是通过其对甲硫氨酸的再甲基化进行的,进而作为SAM形成的底物,或者通过转硫途径代谢。该途径的第一步是HCys与丝氨酸与胱硫醚的缩合反应生成胱硫醚,由胱硫醚β-合酶(CBS)与磷酸吡哆醛(PLP,维生素B6的活性形式)作为辅助因子催化。发现CBS基因的变体(c.844ins68)在甲硫氨酸加载后影响HCys清除率和SAM / SAH比率。c.844ins68也与冠状动脉疾病的风险显着降低有关,但基线HCys水平似乎不受健康个体中CBS基因型的影响。胱硫醚通过PLP依赖性酶胱硫醚 γ-裂解酶(CGL)转化为半胱氨酸,半胱氨酸又与谷氨酸在谷氨酸 - 半胱氨酸连接酶(GCL)的催化下缩合成γ-谷氨酰半胱氨酸。最后,通过谷胱甘肽合成酶(GS)与甘氨酸融合导致谷胱甘肽的形成。 在涉及维生素B12和5-甲基-THF的酶促反应中,甲硫氨酸合酶(MS)促进了HCys的再甲基化。THF通过2个酶促步骤循环回到5-甲基THF,使用维生素B6和黄素腺嘌呤二核苷酸(FAD)作为辅助因子; 甲基最终来源于必需氨基酸丝氨酸。甲硫氨酸的第二种途径是通过甜菜碱同型半胱氨酸甲基转移酶(BHMT)对HCys进行甲基化,其使用甜菜碱作为甲基供体。
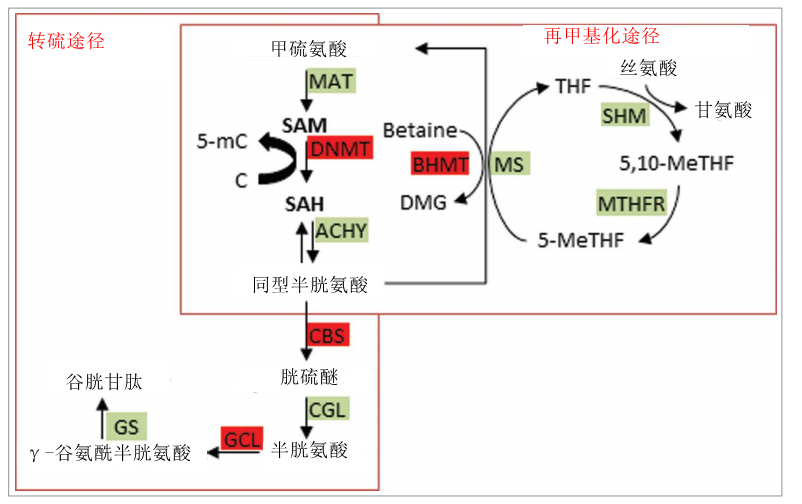
图3. Se干扰一碳代谢。 标有绿色背景是酶; 已经证明受Se影响的酶标有红色背景。 SAM = S-腺苷甲硫氨酸; MAT = 蛋氨酸腺苷转移酶; SAH D S-腺苷高半胱氨酸; DNMT = DNA甲基转移酶; ACHY = S-腺苷-L-高半胱氨酸水解酶; BHMT =甜菜碱同型半胱氨酸甲基转移酶; GCL=D谷氨酸 - 半胱氨酸连接酶; MS = 蛋氨酸合成酶; SHM = 丝氨酸羟甲基转移酶; MTHFR = 亚甲基四氢叶酸还原酶; 5,10-MeTHF = 5,10-亚甲基四氢叶酸; GTK = 谷氨酰胺转氨酶K; AAO = L-氨基酸氧化酶; GR = 谷胱甘肽还原酶; CGL D= 胱硫醚γ-裂解酶; CBS =胱硫醚β-合成酶; SBL =硒代半胱氨酸β-裂解酶。
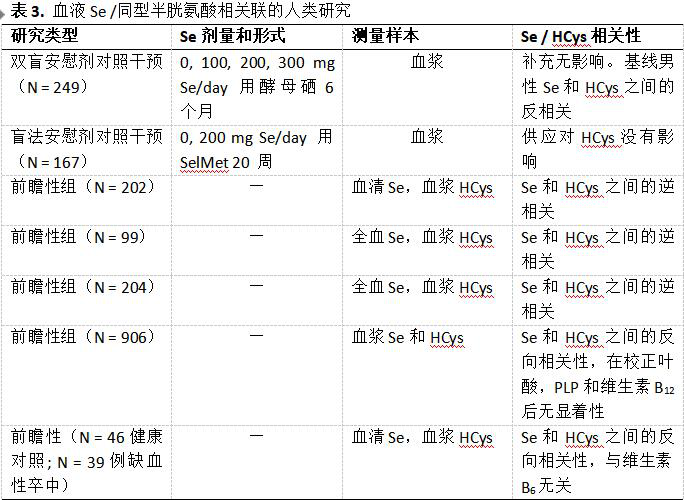
硒状态与血浆同型半胱氨酸水平之间的相关性
一些人体研究显示血浆或血清Se和HCys水平之间呈负相关(表3)。在具有非常高的平均Se状态(635.5mg Se / l血液)的因纽特人群中,硒对血浆HCys呈负性预测。Klapcinska等人在具有低平均Se状态(62.5mg Se / l血液)的群体中类似地检测到全血Se和血浆HCys之间的负相关。虽然两项研究均报告了类似的研究结果,但已知可能影响HCys水平的混杂因素,特别是叶酸和B族维生素,需要加以考虑。在一项研究中,超过65岁的英国国民饮食和营养调查的参与者的Se和HCys之间的反比关系,在调整叶酸,PLP和维生素B12后,这种相关性变得微不足道。然而,在Bekaert等人的研究中,在调整相同的混杂因素之后,Se和HCys的关联仍然很显着,但仅限于男性。通过线性回归分析计算血浆Se占HCys方差的1.8%。与此相符,血清Se预测西班牙人群中HCys的5.8%变异(方差),与叶酸和维生素B12无关,而最高与最低Se三分位组的个体在最高的HCys三分位组中的风险降低63%。年龄小于55岁的缺血性卒中患者在卒中发生不久后(第三天)进行评估,发现其Se水平低于健康对照组,与HCys呈负相关,占HCys方差的15.4%,与维生素B6水平无关。鉴于这些基线关联,已经进行了试验以确定Se补充是否影响血浆HCys。 以每日剂量100或300μg Se作为高Se酵母的形式补充硒6个月增加血浆Se,但对HCys水平没有影响。这种效果的缺乏支持了早期的干预研究,其中每天给予200 μg Se(作为SelMet)20周未能改变血浆HCys水平; 然而,在该研究中未评估Se和Hys在基线处的可能关联。
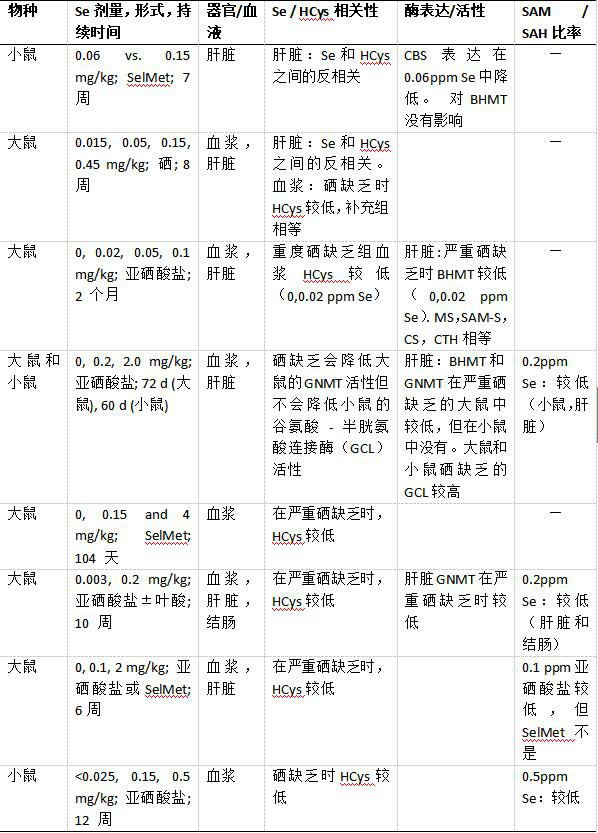
动物实验允许更大的灵活性设计补充研究,更好地控制混杂因素,以及更多的终点检测选项。表4列出了对啮齿动物的研究,饲喂具有不同Se形式和含量的饮食,随后评估了一碳代谢的代谢物和酶。一些与Se的形式和种类无关的一般趋势可以从这些研究中得出:(I)肝脏中似乎存在HCys和Se摄入/状态之间的反相关。这源于对喂食SelMet的小鼠的研究和对喂食硒酸盐的大鼠的研究。两项研究均使用具有可比较Se含量的饮食,这些饮食与人群中存在的膳食摄入量水平相似。这两项研究都使用了与人类饮食摄入水平相当的硒含量的饮食。引人注目的是,与充足的摄入水平(0.15 ppm Se)相比,这两项研究都显示,略微次优的Se摄入水平(0.05 / 0.06 ppm Se)显着增加肝脏HCys浓度,大鼠为30%,小鼠为314%。后一项研究表明,肝脏高同型半胱氨酸血症是由于对CBS的硒依赖性调节所致。其他研究也报道了属于转硫化(GCL)和再甲基化途径(BHMT和甘氨酸N-甲基转移酶)的酶的差异表达,但这些效应似乎是物种特异性的,仅在严重硒缺乏时才能见到。(II)在饲喂被认为缺乏Se(≤0.025ppm)的饮食的那些动物(小鼠和大鼠)中血浆HCys浓度显着降低。(III)在饲喂具有超营养(> 0.2ppm)和足够(0.1-0.2ppm)Se含量的饮食的动物中观察到,血浆HCys水平降低的趋势。
考虑到人类研究显示血浆HCys和Se呈负相关关系,这两个饮食组的比较特别令人感兴趣。由于ACHY催化的反应的可逆性(S-腺苷-L-高半胱氨酸水解酶,参见图3),增加的HCys浓度导致SAH的积累。SAH反过来又是甲基转移酶的竞争性抑制剂,因此,增加的HCys浓度与总DNA低甲基化有关。这一概念得到了一项研究的支持,该研究表明,正常(平均7.2 μM;范围5.8-8.7 μM)HCys血浆水平的女性血浆SAM / SAH比率和淋巴细胞DNA甲基化水平显着高HCys浓度升高(平均12.3 μM; 范围9.3 -16.5 μM)的女性。重要的是,血浆SAH与HCys和淋巴细胞DNA甲基化相关,但SAM不相关,这意味着SAM的测量不足以预测甲基化潜力。还发现组织SAH浓度与小鼠的睾丸,脑和肝中的DNA低甲基化相关。关于鼠Se补充研究中SAH和/或SAM水平的数据是有限且不一致的。三项研究测量了SAM / SAH比率,与缺乏或超营养(2或4 ppm)硒饮食相比,喂养足够(0.1或0.2 ppm)的组中肝脏和结肠SAM / SAH比率较低,然而当使用SelMet代替亚硒酸盐时未见到这一点。另一项研究发现,与0.15和0.025 ppm Se相比,喂食0.5 ppm Se(亚硒酸盐)的小鼠的SAM / SAH较低。最近进行的两项全基因组关联研究发现,位于编码CBS和BHMT的基因附近的SNPs与血液和趾甲Se浓度有关。Combs小组发表的令人兴奋的数据显示干扰一碳代谢会影响硒代谢:ACHY的抑制导致SAM / SAH比率降低,硒转运蛋白SeP从HepG2细胞分泌减少,是由于精氨酸甲基化减少(=更少活性),其对SeP的转录诱导起关键作用。消除硒也需要SAM依赖性甲基化(图1),因此,大鼠中ACHY的抑制导致肝脏和肾脏中的硒滞留,同时减少这些器官和尿液中的排泄性硒形式。这些数据以及动物硒补充研究的结果进一步表明全身硒和一碳代谢是相互联系的。
硒对microRNA表达的调控
通过非编码RNA分子如microRNA(miRNA)靶向mRNA来调节基因表达有时被认为是另外的表观遗传机制。迄今为止,只有一项研究检查了Se是否对microRNA(miRNA)表达有影响。在Se缺陷或Se补充培养基中生长的Caco-2细胞的miRNA谱的微阵列分析(总共737个miRNA),显示12个miRNAs的表达受Se供应的影响。在同一研究中,50种mRNAs的表达水平也是Se反应性的,并且预测许多这些mRNA被Se反应性miRNA靶向。其中之一,miRNA185,其表达在Se缺乏下降低,被证实调节谷胱甘肽过氧化物酶2(GPx2)和硒磷酸合成酶2(SPS-2)的表达。由于SPS-2的酶产物是硒蛋白生物合成机制的一部分,这些发现表明硒可用性部分地影响硒蛋白质组,通过涉及miRNA-185和可能的其他miRNA的表观遗传机制来影响。miRNA-185是Se的一个特别感兴趣的靶标,因为它最近出现了一种肿瘤抑制因子,经常在卵巢癌,乳腺癌,肾癌,前列腺癌和胃癌中被下调,并靶向致癌基因,如Six1,雄激素受体和在半胱天冬酶募集域(ARC)的细胞凋亡抑制因子。硒已被证明在实验环境中以及在某些人体研究中具有抗癌作用(概述); 因此,揭示miRNAs作为硒依赖性肿瘤保护免受恶性转化的介质的可能作用将是未来工作的一个有趣领域。
结论和未来方向
对硒物种引起的表观遗传效应的研究仍然是一个相对较新的领域,尚未得到全面研究。目前主要来自小鼠和基于细胞的数据,以及一些人体研究表明,Se补充和状态修饰全部的和特定基因区域或基因座的DNA甲基化。Se的DNMT抑制及其与一碳代谢的相互作用是发生这种情况的可能途径。另外,Se改变了组蛋白修饰,并且已经显示通过Se代谢产物硒-α-酮酸抑制HDAC活性来发生(至少在体外)。在小鼠和人类干预研究中在小鼠和人类干预研究中,需要系统地评估在全基因组水平上Se相关的对表观遗传标记-DNA甲基化和组蛋白修饰的影响。这些研究将理想地使用不同形式的Se的饮食,具有低初始Se状态的受试者,从而可以观察到剂量 - 反应关系,并且包括转录组评估。联合读数将提供关于Se在表观遗传和转录调控中的作用的重要信息,并利用Se对与疾病风险相关或预测疾病风险相关的表观遗传标记的干扰。这与更好地了解Se在某些疾病(如前列腺癌和2型糖尿病)的预防和进展中的作用特别相关,这些疾病在大规模Se干预试验的不同结果发表后变得不清楚。另一个研究重点是通过Se对DNMT和HDAC酶的抑制进行更深入的了解,以识别活性Se种类、可能的DNMT/HDAC亚型特异性、抑制机制以及体外和体内抑制所需剂量。影响与表观遗传机制相关的核蛋白的Se-反应性信号传导途径,例如通过核小体重塑,转录或DNA修复,如GADD45A所示,也需要进一步检查。我们提出需要对Se引起的表观遗传过程进行详细的全基因组和机制的理解,以完成硒系统生物学的图像,并阐明和预测其对健康结果的影响。
潜在利益冲突的披露
没有披露潜在的利益冲突。
本文系福山生物整理翻译,转载须注明来源自福山生物。